
Meeting Summary
The treatment landscape for patients with multiple myeloma (MM) is constantly evolving. Over the past decade, the introduction of novel agents including proteasome inhibitors (PI) and immunomodulatory agents has led to notable changes in therapeutic strategy and significant improvements in survival. Understanding this landscape and what this means in terms of translating clinical trials to everyday practice is essential.
Prof Paul Richardson opened the symposia with an introduction to currently available agents and recent developments in MM, and highlighted the importance of how we think about current studies. Prof Roman Hájek explored clonal evolution, how it can be prevented in the context of relapsed disease, and the evidence from clinical trials supporting the use of combination therapy. Dr Antonio Palumbo addressed the concept of continuous therapy in MM and where the field is at present. Prof Shaji Kumar described the early phase development of ixazomib. Prof Paul Richardson presented the results from the TOURMALINE-MM1 trial.
Exploring Recent Developments in Multiple Myeloma Therapeutics
Professor Paul Richardson
Prof Richardson opened the symposia by highlighting the array of agents currently available and the recent developments in therapies to treat MM (Figure 1). While the increasing range of effective therapeutic agents is very encouraging and promises to further improve patient outcomes, it adds to the complexity of clinical treatment decisions and clinicians therefore need to carefully examine the evidence base in guiding their decisions. The danger of cross-trial comparison was highlighted by the difference in response rates (RRs) reported in several trials studying the same drug combination in the same disease setting.1,2
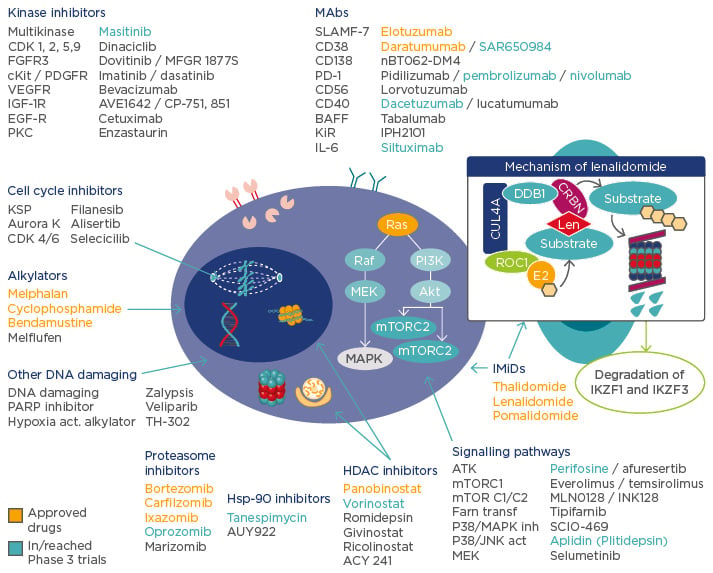
Figure 1: The changing treatment landscape in multiple myeloma.
IMiDs: immunomodulatory drugs; MAbs: monocolonal antibodies; PARP: poly A ribose polymerases; HDAC: histone deacetylase; Hsp-90: heat shock protein 90; IL: interleukin; FGFR3: fibroblast growth factor receptor 3; PDGFR: platelet-derived growth factor receptor; VEGFR: vascular endothelial growth factor receptor; IGF: insulin-like growth factor; EGF: epidermal growth factor; PD-1: programmed cell death protein 1; BAFF: B cell activating factor; KSP: kinesin spindle protein; MAPK: mitogen-activated protein kinase; MTORC: mammalian target of rapamycin complex.
Adapted from Ocio EM et al.79 and Fink EC, Ebert BL.80
Cross-trial comparisons therefore should be approached with caution and it is important to remember that outcomes depend on the patient population and study design which can lead to possible bias and choice of endpoints. Baseline patient characteristics, including cytogenetic risk, International Staging System (ISS) stage, age, renal function, refractory status, and prior therapy, will all influence outcomes and ideally patients baseline characteristics in these parameters should be well-balanced between treatment arms.3-8
Study design should be taken into account when comparing the outcomes of clinical trials; single-arm trials do not adequately characterise time-to-event endpoints (such as overall survival [OS], time-to-progression and progression-free survival [PFS]); therefore, randomised trials are necessary to evaluate these endpoints to account for the variability in the natural history of cancer.9 Open-label trials can lead to assessment bias for PFS.10 Blinding is preferred and is particularly important when patient or investigator assessments are included as components of the progression endpoint.9
Potential sources of bias can arise from the comparison between older studies that used modified European Society for Blood and Marrow Transplantation (EBMT) criteria, which would penalise high rates of complete response (CR) as reappearance of a positive immunofixation electrophoresis would be counted as progression from CR11,12 while more recent trials use international uniform response criteria.12,13 Additionally, open-label studies are at risk of asymmetrical censoring (attrition bias), especially early in the study, which can affect the Kaplan–Meier curve.10 Double-blind, placebo controlled studies, on the other hand, are less vulnerable to bias.
While the ideal main endpoint would be OS, surrogate endpoints (e.g. PFS, RR, and rate of minimal residual disease [MRD]) are commonly used. The depth of response, including MRD, has been shown to correlate with PFS and OS.14-16 Interestingly, outcomes can vary within categories of response; as an example patients who sustain a partial response have longer survival compared with patients achieving and then losing CR17 and patients who improve their response to near-CR (nCR) post-transplant have longer OS compared with patients achieving nCR before transplant.18 Arguably, sustained response may be more important than rapid achievement of deep response.
When interpreting clinical trials, the efficacy (OS, median PFS, hazard ratio [HR] for progression or death, and overall response rate [ORR]), patient population (how many prior lines of therapy), study design, primary endpoints, and eligibility criteria driving the patient study population should be considered. Additionally, when choosing a treatment regimen for an individual patient, the toxicity profile and real-world outcomes need to be considered (such as route of administration, the number of clinic visits, dosing schedule, administration time, whether premedication and pre-hydration are required, and what can be achieved in terms of convenience) (Table 1).
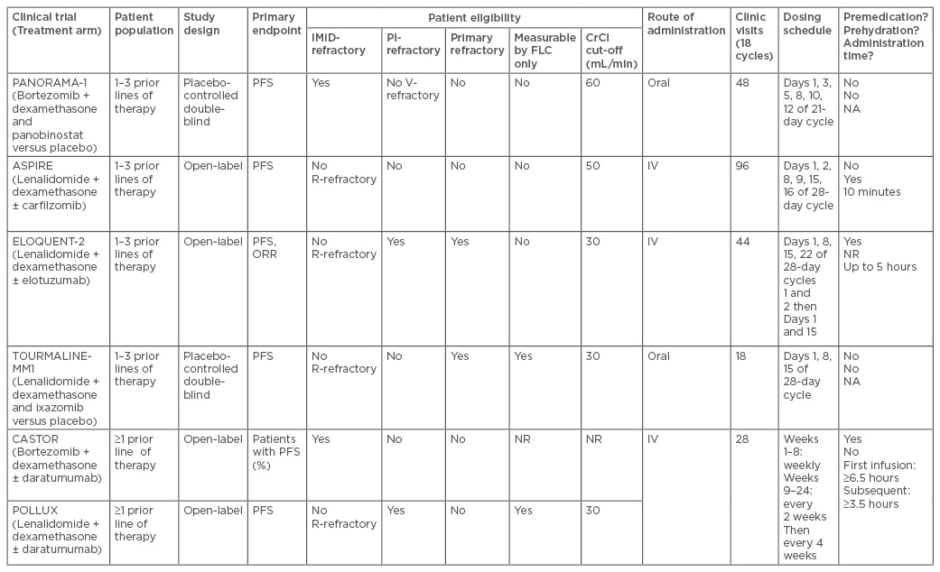
Table 1a: Phase III trials in relapsed/refractory multiple myeloma. Study overview and patient eligibility and real-world considerations.29-34
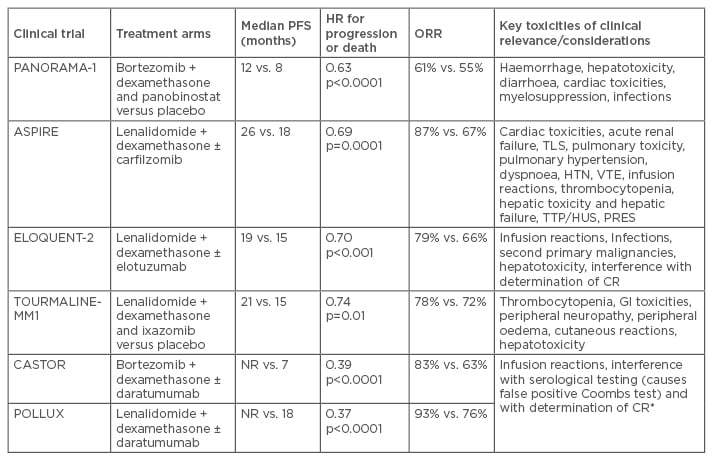
Table 1b: Phase III trials in relapsed/refractory multiple myeloma. Efficacy and key toxicities.
*Based on single-agent studies
IMiD: immunomodulatory drugs; PI: proteasome inhibitors; HR: hazard ratio; ORR: overall response rate; FLC: free light-chain; CrCl: creatinine clearance rate; PFS: progression-free survival; R/V-refractory: lenalidomide/bortozemib refractory; IV: intravenous; N/A: not applicable NR: not reported; HTN: hypertension; TTP: thrombotic thrombocytopenic purpura; HUS: haemolytic-uremic syndrome; PRES: posterior reversible encephalopathy syndrome; VTE: venous thromboembolism; CR: complete response; TLS: tumour lysis syndrome; GI: gastrointestinal.
Preventing Clonal Evolution in Multiple Myeloma: Evidence from Clinical Trials
Professor Roman Hájek
Introducing the concepts of intraclonal heterogeneity and clonal evolution
MM is a complex disease that arises from an initial ‘hit’ that causes premalignant proliferation of plasma cells derived from post-germinal-centre B cells. The natural course of disease evolves from these early stages to monoclonal gammopathy of undetermined significance, progressing to smouldering myeloma, through to active myeloma, and eventually to plasma cell leukaemia, the most aggressive form of disease.19,20
In the initial stages of disease, primary genetic events include immunoglobulin heavy locus (IGH) translocations and hyperdiploidy. Transition throughout different states is thought to involve increasing genetic instability with the acquisition of secondary genetic events, such as copy number abnormalities, DNA hypomethylation, and acquired mutations.20
At the time of diagnosis, multiple genetically distinct subclones are present and evolve over time due to selective pressure imposed by treatment and factors in the microenvironment which can result in disease progression and resistance to treatment.21-25 Myeloma can exhibit either: no heterogeneity (all MM cells are the same), intraclonal heterogeneity (different MM clones that share features), and interclonal heterogeneity (different MM clones that do not share features).25 In a proof-of-concept report, a patient with MM and multiple sites of extramedullary disease was followed serially with biopsies taken from multiple tumour sites. Different mutations were detected in samples from different biopsy sites in the same patient which outlines the concept of spatially divergent clonal evolution.26
What are the clinical implications of clonal evolution?
The clinical implications of clonal evolution can be simplified into three key points: 1) Rationale for earlier therapy when there is less genetic instability and a lower number of clones such as treating high-risk smouldering myeloma;27 2) There is generally no place for monotherapy, targeted therapy, and personalised therapy at the beginning of treatment in both newly diagnosed MM (NDMM) and relapsed/refractory patients, on the contrary combination therapy is needed; 3) However, residual disease may be possibly an important place for the use of targeted therapy.22,25
What are our current strategic approaches?
There are three main therapeutic strategic approaches to address clonal heterogeneity in MM:10,11
- Combining agents with different mechanisms of action (MoAs)
- Sequencing multiple different MoAs
- Prolonging duration of therapy
Combining several drugs with different MoAs has a proven place in preventing clonal evolution, as demonstrated by several clinical trials both in newly diagnosed and in the relapsed setting28-38 that compared doublet versus triplet regimens. All of the reviewed trials demonstrated the superior outcomes seen with triplet over doublet therapies. Furthermore, a meta-analysis of triplet versus doublet regimens in relapsed MM recently demonstrated a clear benefit for triplet therapy in RRs and PFS.39 Among the patients with high-risk cytogenetics, improved outcomes with triplet therapy are particularly notable in TOURMALINE-MM1, where the addition of ixazomib to the lenalidomide-dexamethasone backbone improved median PFS among patients with high-risk cytogenetics; 9.7 months to 21.4 months (HR: 0.54, p=0.02).30 Similarly, in ELOQUENT-2 the addition of elotuzumab to lenalidomide-dexamethasone backbone resulted in improvement in PFS among patients with high-risk cytogenetics (HR: 0.53).31 In newly diagnosed patients, trials comparing triplet and doublet regimens have proven benefits for both PFS and OS outcomes, with impressive results from SWOG S0777, where the combination of bortezomib-lenalidomide-dexamethasone is compared to lenalidomide-dexamethasone. The combination of bortezomib-lenalidomide-dexamethasone, when compared to lenalidomide and dexamethasone, was associated with an improvement of PFS (median PFS: 43 versus 30 months respectively; HR: 0.712; p=0.0018) and OS (median OS: 75 versus 64 months, respectively, HR: 0.709; p=0.025).37
A sequential or alternating MoA schedule was recently investigated by Mateos et al.40 The trial compared a sequential scheme of nine cycles of bortezomib plus melphalan and prednisone (VMP) and then nine cycles of lenalidomide plus low-dose dexamethasone (Rd) or an alternating scheme of VMP and Rd in newly diagnosed transplant- ineligible patients. Compared with treatment with VMP alone or Rd alone reported historically, both alternating and sequential schedules gave a greater RR as well as longer PFS.
In summary, MM is a complex disease, with multiple genetically distinct subclones present at diagnosis.21-24 Intraclonal heterogeneity has implications for disease progression and resistance to therapy through clonal evolution.25 Combining agents with different MoAs may have a synergistic effect and result in a deeper response by targeting all coexisting disease subclones.22,25,41 Sequential use of agents with different MoAs also shows promise in improving outcomes for patients with MM.40,42
Continuous Therapy: Are We There Yet?
Doctor Antonio Palumbo
The progression of myeloma involves increasing genetic and epigenetic abnormalities; and the number of present mutations is increased at each relapse. While patients in the early phase of the disease can expect a 67% likelihood of being progression-free at 5 years, patients who are refractory to prior therapies have a median PFS of around 5 months.43,44 Complete remission in response to therapy is more likely in NDMM than in relapsed/refractory MM.30 Furthermore, with each subsequent relapse up to 40% will not proceed for various reasons to receive subsequent therapy. This is changing the clinical paradigm, where the aim should be to prolong the early phases of disease, while shifting the goals of care to consider early palliative care in the late stages.
Continuous therapy has been developed to prolong the early, sensitive phase of the disease. Although continuous therapy has cost implications, meta-analyses of thalidomide, lenalidomide, and bortezomib maintenance trials show a clear advantage for PFS and OS for maintenance therapy.45
While there may be a theoretical concern about therapy pressure leading to the new treatment-resistant clones, the results of clinical studies to date suggest that continuous treatment does not encourage the development of resistant tumours. On the contrary, when the tumour is not controlled, there is a higher risk of clonal evolution, whereas treatment delays the occurrence of mutations.25 In a pooled analysis of three Phase III trials, an improvement of PFS1, PFS2, and OS was demonstrated for continuous therapy compared with fixed duration therapy, while the duration of second PFS was 15 months with both approaches.
Continuous therapy versus fixed-duration therapy makes a significant difference even in the subgroup of patients who achieved CR in terms of PFS (5-year PFS: 42% and 21%, respectively) and OS (5-year OS: 80% and 54%, respectively).46 The reason for the survival benefit is likely because residual disease is present but undetected, even in patients who appear to have CR.47,48
The question of how long maintenance therapy should be continued has yet to be definitively answered, but tumour reduction can still be seen after 18 months of treatment.49 If possible, treatment should continue until progression. However, continuous therapy must be well tolerated; fatigue, diarrhoea, or other adverse events (AEs) are likely to impact heavily on patient quality of life over 5 years. Toxicities associated with therapies such as bortezomib or lenalidomide, like neutropenia or neuropathy,50-52 will need to be managed or reduced. Early data on ixazomib single agent used in maintenance showed limited peripheral neuropathy and haematologic toxicity which opens the possibilities to examine the use of this PI in the maintenance setting. In fact, there are two ongoing large Phase III studies examining ixazomib for maintenance (TOURMALINE-MM3 and MM4, NCT02181413, and NCT02312258, respectively).
Another PI, carfilzomib, is also being studied in maintenance (FORTE study).53 Maintenance therapy with monoclonal antibodies such as elotuzumab or daratumumab are also being investigated.54
The Ixazomib Development Journey Ixazomib for Myeloma: Where Could it Fit in?
Professor Shaji Kumar
PIs have played a very important role in all stages of myeloma therapy over the past 15 years. PI-based combinations are standard initial therapy regimens, and a meta-analysis has demonstrated improved survival outcomes.55 They are critical for patients with high-risk cytogenetics56 and renal failure,57,58 and have roles in both consolidation and maintenance approaches.49,57 There are some challenges we face with current PIs; most notably, peripheral neuropathy with bortezomib,59,60 potential vascular side effects with carfilzomib,61 and need for parenteral administration with both bortezomib and carfilzomib.62,63 In this context, ixazomib was developed as an oral PI. Preclinically, it was shown to be efficacious against a variety of cell lines as well as xenograft models.64
In clinical trials, ixazomib has shown efficacy as a single agent or in combination.65-68 As a single-agent in Phase I studies of twice-weekly and once-weekly dosing in heavily pretreated MM patients with relapsed/refractory disease, ixazomib achieved ORRs of 15%69 and 27%70 (at maximum tolerated dose), respectively. Its pharmacokinetic properties were not affected by body surface area, enabling fixed dosing of 4 mg (given at Days 1, 8, and 15 of a 28-day cycle), with a reduced dose of 3 mg for patients with severe renal insufficiency or moderate/severe hepatic impairment.71-75
While ixazomib induced a response as a single agent in some patients in early phase clinical trials, combination with dexamethasone improves efficacy. When combined with dexamethasone on an as-needed basis, in patients with relapsed disease who were not refractory to bortezomib, an ORR of about 30%65 was achieved; and in combination with dexamethasone at two different doses, 4 mg and 5.5 mg, ORRs of 31% (95% confidence interval [CI]: 17–49) and 51% (95% CI: 37–71) were achieved, respectively.65 Overall, patients in the ixazomib 5.5 mg treatment arm had more dose reductions, treatment delays, and a higher frequency of AEs (54% of patients in the 5.5 mg arm experienced Grade 3+ AEs compared to 26% in the 4 mg arm) than patients in the 4 mg treatment arm.65
In the newly diagnosed setting, a Phase I/II study of weekly ixazomib in combination with lenalidomide and dexamethasone demonstrated an ORR of 90%, with 27% of patients achieving CR or stringent CR (sCR)66 after 12 cycles. In a similar study with twice-weekly ixazomib dosing, 26% of patients achieved CR, including 19% with sCR.67 When combined with cyclophosphamide and dexamethasone in newly diagnosed patients, the ORR was 77% (95% CI: 63–88). Grade 3 or higher AEs were seen in 73% of patients and most commonly included cytopenias, fatigue, and gastrointestinal side effects.1
Ixazomib’s side effects are generally manageable and toxicities are well managed with dose reduction and supportive care.69,70,75 No cumulative toxicities have been seen within the timeframe of the clinical trials. The most common toxicities include cytopenias (potentially a class effect of PIs), gastrointestinal side effects, and fatigue.69,70,75 Peripheral neuropathy, a significant side effect of bortezomib, does not appear to be very common in single-agent or combination therapy with ixazomib, and when present is generally low grade (Grade 1–2) with infrequent Grade 3 toxicity. Most patients recovered with dose modification.
Ixazomib has been shown to be effective with manageable side effects and, as an oral drug, is potentially more convenient. It is currently being studied in combination with alkylators such as melphalan and cyclophosphamide, immunomodulatory drugs, histone deacetylase inhibitors, and monoclonal antibodies. Future directions aim to explore suitability with other novel drugs and alternating dosing schedules.
The Phase III TOURMALINE-MM1 Study
Professor Paul Richardson
As has been discussed in this symposium review, PIs form the backbone of therapy29,44,67,76,77 but the critical point is the shift in treatment patterns towards extended therapy. In that context, acceptable side effect profiles are key to enable patients to continue therapy for prolonged periods of time.77 Preclinical studies of ixazomib and lenalidomide have indicated synergy64 and early-phase studies of ixazomib-Rd in NDMM appeared to be promising;66,67 providing a rationale for the Phase III TOURMALINE-MM1 study.30
The TOURMALINE-MM1 study was the first Phase III study of an all-oral triplet regimen incorporating an oral PI and an immunomodulatory agent in relapsed/refractory myeloma patients.30 After the Phase I development of ixazomib defined an optimal dose and schedule,68,69 combination studies in the newly diagnosed setting appeared to support the use of Ixazomib in combination with lenalidomide.65
Study design
The global, double-blind, placebo-controlled study enrolled 722 patients with relapsed/refractory MM who had received 1–3 prior lines of therapy. Patients in the investigational arm (n=360) received ixazomib 4 mg on Days 1, 8, and 15, lenalidomide 25 mg on Days 1–21 and dexamethasone 40 mg on Days 1, 8, 15, and 22 (IRd). Patients in the control arm (n=362) received placebo, lenalidomide, and dexamethasone (placebo-Rd).30
Key inclusion criteria included confirmed diagnosis of MM, measurable disease by serum protein electrophoresis, urine protein electrophoresis or free light-chain assay, 1–3 prior lines of therapy, relapsed and/or refractory disease and creatinine clearance ≥30 mL/min. Patients who were refractory to previous PI or lenalidomide-based treatment and those with peripheral neuropathy higher than Grade 1 with pain or Grade 2 or more were excluded.30
Patient demographics were well balanced across both groups, including median age, ISS stage, prior lines of therapy, PI exposure, type of prior regimen, and cytogenetics.
Key efficacy data
The median PFS (primary endpoint) at the final PFS analysis was 20.6 months in the IRd arm and 14.7 months in the placebo-Rd arm (p=0.012, HR: 0.74). While not statistically powered for PFS subgroup analysis, PFS benefit was consistent across key pre-specified patient subgroups, including those with poor prognosis, such as patients with high-risk cytogenetic abnormalities (defined as del(17p), t(4;14) or t(14;16)), multiple prior lines of therapy and high disease burden. The median PFS data indicate that an ixazomib regimen might offer improved prognosis for patients with high-risk cytogenetics by lengthening PFS to a value that is similar to that among patients with standard-risk cytogenetics. Among the patients with high-risk cytogenetics, median PFS for IRd and placebo-Rd was 21.4 months and 9.7 months, respectively (HR: 0.543). There was an 11-month median PFS benefit for patients with del(17p) and a 7-month median PFS benefit for patients with t(4;14) translocation. Median OS was not reached in either arm. IRd improved RRs versus placebo-Rd, and deepening responses were observed with ongoing treatment.30
Key safety data
In terms of tolerability, the median relative dose intensity was 97.4% and 98.8% for ixazomib versus placebo. There was a higher frequency of Grade ≥3 AEs with IRd versus placebo-Rd, primarily due to thrombocytopenia. Serious AEs were marginally higher in the placebo group (49% in placebo-Rd versus 47% in IRd). In general, the difference in AEs was not pronounced; although, one AE, rash, was more prominent in the treatment group, from clinical experience, this is generally manageable.30 Importantly, patient-reported quality of life (as measured by EORTC-QLQ-C30) was maintained when ixazomib was added to the lenalidomide/dexamethasone doublet regimen.30,78
Overall, ixazomib, when combined with Rd for patients with relapsed or refractory MM and compared with placebo-Rd, was associated with a significant and clinically meaningful improvement in PFS, improved time-to-progression and RRs.While not powered for subgroup analysis ixazomib regimen improved PFS in high-risk patients that was similar to that seen in the overall patient population and standard-risk patients. Limited additional toxicities were seen with ixazomib; and importantly, rates of peripheral neuropathy, cardiac, or renal AEs were low, making long-term treatment feasible.30 Ixazomib has since been approved in the USA in combination with lenalidomide and dexamethasone for the treatment of patients with MM who have received at least one prior therapy.74





