Meeting Summary
Haemophilia A (HA), defined by factor VIII (FVIII) levels ≤40 IU/dL, is a chronic condition with consequences beyond bleeding complications. Jan Astermark (Professor of Clinical Coagulation Medicine, Senior Consultant, and Head of Department of Translational Medicine, Lund University; and Department of Haematology, Oncology and Radiation Physics, Skåne University Hospital, Malmö, Sweden) outlined the burden of HA on the quality of life (QoL) of patients, including bleeding, joint damage, pain, psychosocial wellbeing, and physical activity. He shared real-world evidence showing that current prophylactic regimens with FVIII or non-factor therapy (NFT) are not sufficient to eliminate all types of bleeds and that many challenges remain. Astermark presented several analyses highlighting that FVIII levels in the non-haemophilia range may be necessary to prevent residual bleeding. These analyses have informed recent treatment goals that transcend historical targets of converting severe HA (SHA) into moderate or mild forms and aim towards normalised haemostasis to eliminate bleeds. Maria Elisa Mancuso (Senior Consultant in Haematology, Centre for Thrombosis and Haemorrhagic Diseases, IRCCS Humanitas Research Hospital and Humanitas University, Milan, Italy) presented a patient case to illustrate the challenges that people with HA (PwHA) face over their lives and the evolution of treatment strategies to address unmet needs. Christoph Königs (Head of Clinical and Molecular Haemostasis at the Department of Paediatrics and Adolescent Medicine, Clinical and Molecular Haemostasis, Goethe University, University Hospital, Frankfurt, Germany) emphasised the unique challenges faced by children with HA and their caregivers, including restrictions in daily activities, regular evaluations for subclinical and evident bleeds, long-term joint protection, delayed inhibitor development, self-injection skills, and suboptimal adherence. He discussed how standard and extended half-life (SHL, EHL) therapies have improved care in children with HA but highlighted how prophylaxis with existing therapies is not sufficient to eliminate evident and subclinical bleeds. He concluded by sharing data on novel therapies that offer the potential to maintain FVIII levels in the non-haemophilia range (≥40 IU/dL) to help address these unmet needs. Robert Klamroth (Head of the Department of Internal Medicine, Vascular Medicine and Haematology, and Director of Haemophilia Treatment Centre, at Vivantes Klinikum Friedrichshain, Berlin, Germany) focused on the evolving challenges of HA in adulthood, including surgery and the need for anticoagulant or antiplatelet therapy for the management of comorbidities. Recent clinical data were shown to demonstrate how high sustained FVIII levels could minimise bleeding risk and improve joint health, surgical management, and overall QoL in adults with HA. In the panel discussion, two patient cases were reviewed to consider unmet needs in people with mild HA and in elderly people with HA, and the panel summarised how sustaining FVIII levels in the non-haemophilia range could help address these needs. The panel concluded by reviewing the evolution of treatment strategies and the importance of targeting normalised haemostasis in a new era of protection in HA.
Welcome and Introduction
Jan Astermark
Despite regular and early prophylactic treatment, PwHA at all ages still experience bleeds that impact their daily lives. In a real-world cross-sectional survey of 244 people with severe or moderate HA treated with prophylaxis in the USA, 25% of patients had joint problems and 43% experienced at least one bleed in the year prior to the survey.1 Bleeding and joint problems, in turn, have a significant burden on patients’ QoL, with 39% of patients experiencing disruptions to daily living, 28% being unable to work full time, and 84% sometimes or often avoiding physical activity because of HA.1
Recent analyses indicate that sustained time in the non-haemophilia range (FVIII levels ≥40 IU/dL) may be a necessary first step to elevating the standard of care in PwHA (Figure 1).2-6 Targeting FVIII levels ≥40 IU/dL could improve clinical outcomes and the QoL of PwHA by reducing the risk of clinical and subclinical bleeds, improving long-term joint health, and reducing synovitis, joint damage, and pain.2-5 This symposium discussed this new era in HA protection and explored how novel therapies may help achieve normalised haemostasis to improve the physical and mental well-being of PwHA.
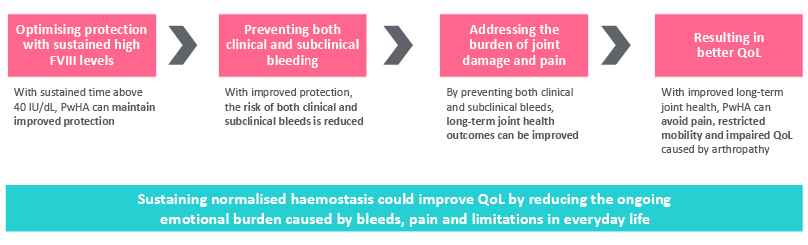
Figure 1: Normalisation of haemostasis as the beginning of the journey to elevate standards of care.2-6
FVIII: factor VIII; PwHA: people with haemophilia A; QoL: quality of life.
Evolving Goals in Haemophilia A: From Concept to Reality
Maria Elisa Mancuso
Mancuso illustrated the challenges associated with HA and the evolution of treatment strategies via the example case of a male patient who was diagnosed with SHA at 14 months old following a gluteus haematoma.
Childhood
To manage the gluteus haematoma, the patient was treated with SHL-FVIII and started on regular prophylaxis to minimise the risk of future bleeds. In children, venous access can be a challenge; for this child, it led to skipped infusions and suboptimal protection. He experienced recurrent joint bleeds and so began prolonged treatment with a central venous line to help optimise prophylaxis. At 8 years old, he experienced a central venous line infection that necessitated a temporary stop in SHL-FVIII, but his regular treatment was resumed once the infection subsided.
Adolescence
When he was 11 years old, the first EHL-FVIII became available, but his parents were reluctant to switch his treatment in fear of inhibitor development. He continued on SHL-FVIII for a further 7 years until, after recurrent target joint bleeds, he decided to switch to EHL recombinant FVIII (rFVIII) at 15 years old.
When playing basketball at school, he noticed mild ankle discomfort that he managed with injections prior to training. However, an ultrasound evaluation identified synovial hypertrophy at his right ankle and left elbow as well as initial chondral changes at his left knee. His trough level at 72 hours was 3–6%. To his disappointment, he was advised to change hobby.
Adulthood
The patient continued his treatment of EHL-rFVIII but variable venous access and a reluctance to inject at inconvenient times resulted in suboptimal adherence. To minimise treatment burden, he switched to a once-weekly NFT, which lessened his joint pain to a degree that he felt able to return to the gym. However, over the next few years, he experienced several spontaneous bleeds in his left knee, and subsequent ultrasound evaluations showed synovial hypertrophy at his right ankle and evidence of progressive osteochondral changes at his left knee. He decided to switch back to EHL-rFVIII to intensify his prophylaxis and started selective Cox-2 inhibitors and regular physiotherapy to help manage his pain.
Ageing
Despite his treatment programme, his knee pain continued, and the patient was indicated for knee replacement surgery when he was 42 years old. The surgery was successfully managed with perioperative FVIII management, and he subsequently entered a trial with a rebalancing agent administered once every 2 months. This regimen minimised his treatment burden, but ongoing pain limited his activity leading to gradual weight gain and the development of hypertension and mild dyslipidaemia. In his sixties, he underwent surgery for prostate cancer that necessitated antagonism of the NFT effect. EHL-rFVIII was used and FVIII activity levels were closely monitored to ensure adequate haemostasis during and after his prostatectomy.
Mancuso concluded her presentation by summarising the evolution of treatment strategies. Treatments for HA historically focused on converting severe disease (FVIII <1 IU/dL) into moderate forms (FVIII ~1–5 IU/dL) to minimise life-threatening bleeds. Improvements in care were observed with EHL therapies and NFTs; however, these are not sufficient to fully protect patients from bleeds, including subclinical and joint bleeds.7 With the development of novel treatment options, including ultra-long FVIII replacement therapy, it is feasible to now target more ambitious goals of normalised haemostasis.2
Realising New Possibilities for Children with Haemophilia A
Christoph Königs
Königs emphasised the unique challenges faced by children with HA and their caregivers, including the risk of inhibitor development, lack of self-injection skills, suboptimal adherence, the need for regular evaluations of subclinical and joint bleeds, and limitations in physical activities (Figure 2).
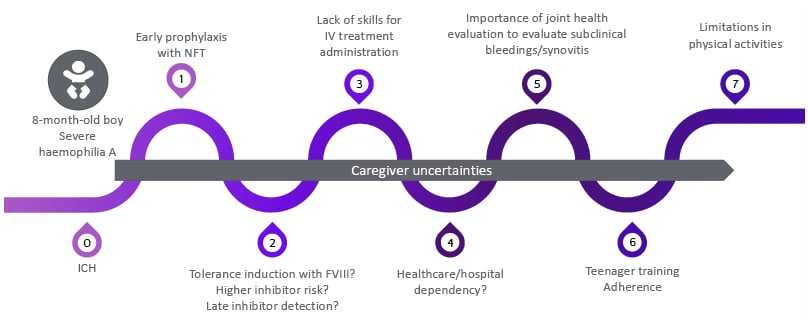
Figure 2: Challenges for children with SHA and their caregivers.10,20-26
FVIII: factor VIII; ICH: intracranial haemorrhage; IV: intravenous; NFT: non-factor therapy; SHA: severe haemophilia A.
Children with HA can be affected by significant joint and muscle bleeding. In a UK cohort of 237 PwHA (including 80 children <18 years) selected for high compliance with EHL or SHL prophylaxis, 32.5% were affected by joint bleeds.8 Similarly, in a real-world, retrospective, multi-institutional cohort of 314 children and young adults with HA receiving NFT, 15 patients (including four with inhibitors) experienced 19 muscle bleeds that required intensive and prolonged factor treatment.9 Impaired joint health in the absence of reported bleeding was also documented in a cross-sectional study of 106 children (636 joints) with SHA on prophylaxis, where 50% scored >0 in Hemophilia Early Arthopathy Detection with Ultrasound (HEAD-US) and 13% of joints with no bleeding history were scored ≥1 in HEAD-US.10
Among the current treatment approaches for children, emicizumab is an NFT indicated in the European Union as routine prophylaxis in PwHA of all ages with FVIII inhibitors and in those without FVIII inhibitors if they have SHA or moderate HA with a severe bleeding phenotype.11 In the Phase IIIb HAVEN 7 trial, emicizumab prophylaxis demonstrated efficacy and a favourable safety profile in 55 infants aged ≤1 year with SHA without inhibitors.12 After a median efficacy period of 101.9 weeks, the model-based mean (95% CI) annualised bleeding rate (ABR) was 0.40 (0.30–0.63) for treated bleeds, and 54.5% of patients experienced no treated bleeds.12 Emicizumab had an acceptable safety profile with no intracranial haemorrhages or new safety signals identified during the study.12 Prior studies have reported that emicizumab maintains FVIII-equivalent levels in animal models of ~9–20 IU/dL and FVIII-like activity in clinical trials of ~15–20 IU/dL.13-16 However, FVIII-like activity reported for PwHA treated with emicizumab cannot be compared with, or interpreted as equivalent to, FVIII activity reported in participants treated with FVIII.12,14
Advances to sustain FVIII levels within the non-haemophilia range could help to prevent evident and subclinical bleeding in PwHA. Efanesoctocog alfa is an FVIII replacement therapy indicated in the European Union for the treatment and prophylaxis of bleeding in PwHA of all ages.17 In the Phase III XTEND-Kids trial in people with SHA (PwSHA) <12 years, efanesoctocog alfa provided high sustained FVIII activity in the non-haemophilia range (>40 IU/dL) for 3 days and >10 IU/dL for almost 7 days after administration, leading to effective bleeding treatment and prevention.18 The mean (95% CI) ABR for treated bleeds was 0.61 (0.42–0.90) for overall bleeding and 0.16 (0.0–0.30) for spontaneous bleeding (sensitivity population; n=73), and 88% of participants had zero treated spontaneous bleeds.18 Efanesoctocog alfa was well tolerated, with no adverse events leading to discontinuation. No inhibitors or antidrug antibodies were reported.18 Low bleed rates were maintained for children (n=71) throughout the XTEND-ed extension study, where over a median (range) cumulative treatment duration of 119.1 (88.1–152.6) weeks with efanesoctocog alfa, a mean (95% CI) ABR of 0.67 (0.48–0.93), and 83% (44/53) of patients with zero bleeds at Months 12–18 were reported.19
Achieving sustained FVIII levels in the non-haemophilia range makes the ambition of a haemophilia-free mind for children with HA and their caregivers more feasible.2,5
Transforming Clinical Perspectives in Adults with Haemophilia A
Robert Klamroth
Adults living with SHA face multiple challenges, including healthcare dependency, evident and subclinical bleeds, physical activity limitations, surgeries, and comorbidities requiring concomitant medications, including anticoagulants and antiplatelets.9,20,26-36
Pain, particularly joint pain, is a significant burden to PwHA and is often associated with a higher use of pain, depression, and anxiety medications compared with population controls.37,38 In a German survey including adults with HA (N=513), pain prevalence increased with age, and over two-thirds of patients aged >40 years reported frequent joint pain.38 In a Phase III, open-label, multicentre study of PwSHA, patients reporting no pain were significantly more likely to have zero bleeds (57%) than patients reporting pain (43%; p<0.05).39
Despite prophylaxis, adults with HA can still experience bleeding, including spontaneous joint bleeds. In a 12-month observational study in the UK including 237 PwSHA (157 adults), 60% of adults were affected by joint bleeds despite FVIII prophylaxis.8 Similarly, in an observational cohort of 40 PwSHA in Italy, 25% developed spontaneous joint bleeding despite receiving NFT. In the latter study, thrombin generation assays failed to differentiate patients by risk of spontaneous joint bleeding, but synovitis and higher HEAD-US score were strong predictive factors for spontaneous joint bleeds.28
Targeting FVIII levels in the non-haemophilia range is supported by data from a Phase III gene therapy trial, which found that 100% of patients (n=11) with FVIII levels ≥40 IU/dL had zero treated bleeds after Year 2 (Figure 3).40 Once-weekly efanesoctocog alfa provides high sustained FVIII activity, with a mean steady-state half-life (CI) of 47.0 (42.3–52.2) hours, thus enabling FVIII levels ≥40% for 4 days and >15% for 7 days in adults.41 In the first 2 years of the XTEND-ed extension trial, efanesoctocog alfa demonstrated sustained bleed protection in adults and adolescents (N=146), with a mean (standard deviation [SD]) overall ABR of 0.64 (1.0) and mean (SD) spontaneous ABR of 0.24 (0.5), after a median (range) cumulative treatment duration of 170.5 (46.3–192.6) weeks.41,42 The proportion of patients with zero bleed rates remained high and stable over the full efficacy period, from 74.3–80.4% for overall bleeds and from 87.5–93.2% for spontaneous bleeds.41,42 Improvements were also observed in haemophilia joint health scores (particularly in patients >50 years) and Haem-A-QoL (mean [SD] change in physical health: -7.5 [20.05]; total score: -5.8 [13.35]).41-43 Efanesoctocog was overall well tolerated. No FVIII inhibitors were detected. Two patients experienced thromboembolic events: one was a deep vein thrombosis following corrective surgery for a femur fracture (in the setting of treatment with another FVIII product), and the other was a cerebral infarction in a patient with pre-existing atrial fibrillation and other risk factors; neither event was related to efanesoctocog alfa treatment. Two related treatment-emergent adverse events were reported (facial paralysis and locally measured coagulation FVIII level decrease), and both events resolved.42,44
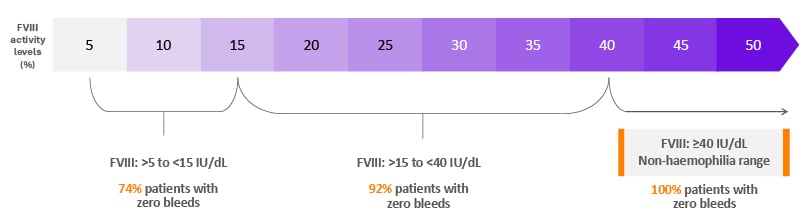
Figure 3: Maintaining FVIII levels in the non-haemophilia range could help achieve the goal of zero bleeds.40
FVIII: factor VIII.
In PwHA, surgery requires careful management of bleeding risk by maintaining high perioperative FVIII levels. Over 4 years in the XTEND clinical programme, 45 patients underwent 62 major surgeries (31 orthopaedic). A good or excellent haemostasis response was reported in 95.2% of patients, with low mean (SD) perioperative efanesoctocog alfa consumption (Days -1 to 14) of 176.2 (54.6) IU/kg across an average (SD) of 4.1 (1.7) injections.45 Few injections and low perioperative consumption was also observed for minor surgeries, of which 47 patients (56 surgeries) had a mean (SD) perioperative consumption (Days -1 to 7) of 94.3 (31.8) IU/kg, across an average (SD) of 1.9 (0.8) injections.45
Transitioning From Clinical Trials to Real-World Practice
Speaker presentations were followed by a panel discussion of two patient cases to demonstrate the challenges of PwHA.
The first patient was a 15-year-old boy living with mild HA. He was receiving on-demand EHL-rFVIII that fit well around his active lifestyle and had no recent history of joint bleeds or obvious symptoms of his disease. In a routine ultrasound, however, he was found to have synovitis in both ankle joints. In an audience poll, long-term joint health and the ability to be physically active/play with friends were voted to be the most important outcomes to consider for a child/adolescent like this patient. The panel agreed that regular prophylaxis was important to maintain high FVIII levels, even in patients living with mild HA. Furthermore, despite low evident bleed rates, it is important to maintain optimal adherence and regular ultrasound monitoring to ensure adolescents do not experience clinical or subclinical bleeds that could contribute to long-term joint damage.2,4,46
The second case introduced a 77-year-old man living with SHA and concomitant cardiovascular disease. In an audience poll, approximately half of the audience thought the optimal required FVIII level to permit dual antiplatelet therapy without bleeding risk in PwHA was 20%, with approximately one-third choosing FVIII levels of 30%. Based on the current available recommendation, the panel agreed on a baseline FVIII coagulation >20% for dual antiplatelet therapy or oral anticoagulation, and recommendations suggest the duration of such therapies should be as short as possible.35,47
The panel concluded that treatment goals for PwHA are evolving to optimise health and well-being.3,7 Haemophilia therapies that can sustain FVIII levels in the non-haemophilia range could help overcome the challenges that children and adults with HA face and improve QoL by reducing the emotional burden caused by bleeds, pain, and limitations in everyday life.3,4,6,48
NP-39929
March 2025





