Meeting Summary
Prof Robin Foà opened the symposium by highlighting how improving healthcare and an ageing population are increasing the burden on healthcare resources and creating challenges in maintaining the high level of healthcare provision that many people expect. Dr Armando López-Guillermo discussed the role of biosimilars in maintaining sustainable and affordable healthcare systems and the need to balance this against ensuring that biosimilars offer comparable efficacy and safety compared with their reference products. Dr Martin Schiestl outlined the differences in approval processes for biosimilars compared with novel biological therapies and generic versions of small-molecule drugs, and how this ensures similarity between biosimilars and their reference products. Prof Steffen Thirstrup reviewed the processes that European Union regulatory authorities undertake when deciding whether it is appropriate to extrapolate indications for biosimilars beyond a single approved indication. The meeting objectives were to discuss the role of biosimilars in meeting healthcare needs and to review what regulatory assessments biosimilars undergo prior to receiving marketing approval, and how additional extrapolated indications can be scientifically justified.
Chairperson’s Introduction: How Can We Sustain Healthcare Provision?
Professor Robin Foà
Ageing societies are placing an increasing burden on healthcare resources, not only in terms of a greater number of people living longer, but declining birth rates are also resulting in populations composed of a higher proportion of older people than in the past. However, the ‘biological age’ of patients is also younger than it once was. For example, anyone who has lived to 70 years old can now be expected to live for another 20–25 years and these people want to maintain their quality of life as they age, which potentially means living with what may have once been a life-threatening disease that can now be managed as a chronic disease.
Many of these diseases are managed with biological therapies that have been driven by genetic technologies that evolved from generating simple peptides in bacteria and yeasts, such as aspirin, to producing complex glycoproteins, such as monoclonal antibodies using mammalian systems. However, the costs associated with new treatments are ever-increasing and risk limiting access to treatment (Figure 1).1 Accordingly, biosimilars, biologicals that have been shown to be highly similar to the originator or reference product in terms of the requirements for comparability, may play a role in improving access to biological treatments.

Figure 1: The challenge in sustaining healthcare provision.
The Need for Biosimilars and the Challenge of Extrapolation: A Clinical Perspective?
Doctor Armando López-Guillermo
Biologicals have revolutionised the treatment options for many disabling and life-threatening diseases, including kidney disease, cancer, arthritis, psoriasis, and growth disorders.2-6 The advantages of biologicals have been exemplified in haematology, where the addition of rituximab to chemotherapy in patients with B lymphoproliferative disorders has significantly improved overall survival.7-13 Therefore, although the incidence of T cell lymphatic disorders has likely increased over the last decade, the incidence of death has decreased, which can most likely be attributed to the introduction of rituximab immunotherapy over the past few decades.14
However, a major limitation to the use of biologicals is their expense; biologicals account for <1% of all prescriptions, but up to 28% of prescription drug costs.15 The rise in long-term chronic conditions treated with biologicals, alongside increasing patient expectations among ageing populations means that providing affordable and sustainable healthcare to these patients is becoming increasingly challenging.
Biosimilars may increase access to biological treatments by offering a more sustainable and affordable treatment option. Despite the affordability and comparable efficacy of biosimilars to the reference product, a primary barrier to their widespread use is clinician concerns regarding product quality, the lack of clinical and safety data, interchangeability, and extrapolated indications.16 Although biosimilars are not exact replicas of the reference product, clinicians should understand that, before receiving marketing approval, biosimilars must undergo robust regulatory assessments and demonstrate clinical efficacy, safety, and product quality that is comparable with the original product.
Due to the complexity of monoclonal antibodies, it is not possible to generate an exact replica of a biological product.17,18 Therefore, the goal of biosimilar development is to create a replica that is as close to the reference product as possible with no clinically meaningful differences in pharmacokinetics, pharmacodynamics, efficacy, safety, and immunogenicity.19 To achieve this, testing of biosimilar monoclonal antibodies relies on the well-established principles of having:19
- An identical amino-acid sequence to the original product
- High similarity in the chemical and biophysical characteristics of the original product
Following confirmation of similarity, clinical efficacy and safety must be validated in head-to-head trials with the reference product, which are used by the European Medicines Agency (EMA) to assess whether there are any clinically significant differences between the biosimilar and the reference product. To achieve this, validation studies must be conducted using a (preferably) double-blind, randomised, controlled trial that is adequately powered to assess non-inferiority.20 For these studies, the most sensitive, homogenous, immunocompetent patient population should be used to detect any small product-related differences, minimise variability, and accurately assess immunogenicity.21 Trial data for rituximab, for example, suggest that the largest effect size is seen in patients with follicular lymphoma treated with a rituximab-cyclophosphamide-vincristine-prednisone combination, making this a sensitive population for testing rituximab biosimilars.9,22-25
Provided a biosimilar has proven efficacious in a single, sensitive indication of the reference product, extrapolation to other approved indications of the reference product may also be justified. When considering extrapolating indications for a biosimilar, clinical experience in the indications of interest and the characteristics of the functional moieties of the biosimilar are considered against those of the reference product.26 Additionally, the biosimilar must have the same mechanism of action for each indication as the reference product,26 so obtaining an extrapolated indication for monoclonal antibodies with multimodal mechanisms of action can be difficult. For example, extrapolating the indication of rituximab biosimilars is challenging because different polymorphisms and mutations in B cell malignancies can influence CD20 expression, which in turn can affect affinity and response to rituximab.27 It is also unclear whether the mechanism of action of rituximab varies depending on the lymphoma subtype or localisation. Therefore, these concerns are valid considering that there are differences in CD20 expression in vitro, and rituximab dose adjustment is required for different B cell malignancies, tumour burden, and phenotypes in the clinical setting.27,28 Additionally, it is unclear whether the mechanism of action of rituximab remains the same when used in combination with chemotherapy. Therefore, in these cases additional clinical evidence may be required to support extrapolating the efficacy and safety of a rituximab biosimilar beyond its initial indication.
In conclusion, ageing populations and an increasing number of patients with chronic disease are driving a desire for increased access to biological therapies; biosimilars offer a more affordable treatment option that can increase patient access to high-quality, clinically effective therapies. Biosimilars must meet the same quality standards as novel biologicals and undergo rigorous testing before approval is granted from the EMA or US Food and Drug Administration (FDA). Although there are concerns regarding extrapolating indications beyond a single indication, each indication is systematically evaluated independently and, in the case of monoclonal antibodies with multimodal mechanisms of action, further clinical evidence is often required to justify extrapolation.
The Science of Biosimilars: From Quality to Extrapolation
Doctor Martin Schiestl
Extrapolating indications for biosimilars remains controversial among clinicians. Despite regulatory bodies such as the EMA and FDA thoroughly evaluating the evidence for and against extrapolation prior to approval, some medical groups have critiqued the decisions of the EMA and expressed uncertainty about the findings, and have therefore recommended against using biosimilars in extrapolated indications.29-31 In particular, clinician uncertainty is likely to be related to queries regarding the science behind the regulatory approval pathway for biosimilars and the way these products are developed.
Clinicians are generally familiar with the development of new products, which focusses on the clinical evidence demonstrating safety and efficacy in various indications. However, the goal in biosimilar development is to demonstrate similarity of a product containing essentially the same active ingredient as the reference product,32 and having no clinically meaningful differences,33 so that the clinical experience of the reference product can be applied to the biosimilar.
The most sensitive tools for demonstrating similarity are analytical methods for measuring the physicochemical and functional properties of products. Therefore, the analytical comparison sets the foundation for demonstrating biosimilarity, which is complemented by non-clinical and clinical confirmation of similarity (Figure 2).
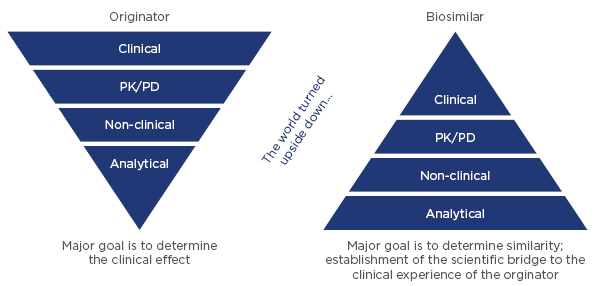
Figure 2: Comparison of the development approach for originator biologicals and biosimilars.
PD: pharmacodynamics; PK: pharmacokinetics.
For a biosimilar to receive marketing approval, the primary amino-acid sequence and protein folding must be identical to the reference product.34 Other features, including post-translational modifications such as glycosylation, are required to have high similarity. However, differences are acceptable provided there is evidence to show that any differences are not clinically relevant, and it is important to note that protein glycosylation also varies from batch to batch for reference products, which is considered to be normal and is usually not problematic, provided this variability is controlled within acceptable margins.34,35 Additionally, following approval of a reference product, it is common for the manufacturing process to be changed from time to time; for example, the cell culture media, purification methods, or manufacturing sites may be changed.36 These changes are tightly controlled by regulatory authorities and are only approved when there is evidence that they do not result in clinically meaningful differences to the product.37 This evidence is also based on analytical comparisons, and may be supplemented by non-clinical, or very rarely, clinical confirmation, which is then extrapolated to all indications. Therefore, the concept of extrapolation of indications for biosimilars on the basis of overall comparability is not new, given that it is already being continuously applied to reference products.
However, based on the complexity of proteins, and the many structural and functional attributes (i.e. quality attributes) that need to be compared, there is concern that minor, but clinically relevant, product differences may be overlooked. To address these concerns, each quality attribute is assessed systematically for its contribution to immunogenicity, toxicity, pharmacokinetics, and efficacy. The clinical relevance of many quality attributes of biologicals are well understood and can be assessed independently for each indication.38-49 For example, it is well known that amino-acid sequence, protein folding, and glycosylation influence efficacy, whereas attributes such as amino-acid sequence, aggregates, host-cell proteins, and certain glycans can affect the immunogenicity of biologicals.38-49 By understanding these features and comparing those of a biosimilar with the reference product, efficacy and immunogenicity can be controlled so that they are comparable.
Functional characterisation is also a key component in the biosimilar exercise and for elucidating structure–function relationships to increase product understanding.50 The availability of precise bioassays allows assessment of the functional properties of biologicals and to evaluate the potential clinical impact of minor differences in certain quality attributes between the biosimilar and reference product, if they exist.50
As extrapolation to other indications relies on the principle of sameness between the biosimilar and the reference product, each extrapolated indication is independently evaluated on the basis of totality of available evidence and is not automatically granted for all indications of the reference product.20,34 An extrapolated indication for a biosimilar must therefore be justified on the basis of the understanding of the mechanism of action, and the comparative analytical, non-clinical, and clinical data between the biosimilar and the reference product.16,26
To conclude, for biosimilars to be approved they must undergo rigorous analytical, non-clinical, and clinical testing to establish similarity with the reference product. An extrapolated indication is systematically and independently evaluated and approved based on the totality of evidence for the biosimilar compared with the reference product.
The Evidence for Extrapolation: A Regulatory Perspective
Professor Steffen Thirstrup
As recently as 2014, both the EMA and the FDA have issued guidance on the evidence required for marketing authorisation for biosimilars,20,34 but key regulatory requirements for assessing products, whether reference biologicals, biosimilars, or small-molecule generics, have not changed.50 However, the type and/or amount of data required for marketing authorisation varies, depending on the type of product. The documentation required for approving a new product is considerable and extensive data documenting the product’s quality (manufacturing, stability, and its controls), non-clinical safety, as well as clinical efficacy and safety must be provided for agency review. In the case of biosimilars, in addition to establishing quality, as for any other product, the manufacturer must also establish biosimilarity to the reference product in all aspects, that is, the product is as similar as current methods allow. Furthermore, clinical data must be provided in at least one indication that establishes comparable efficacy and safety with that of the reference product using sensitive endpoints. In contrast, small-molecule generics are only required to demonstrate equivalent bioavailability (bioequivalence) to claim therapeutic equivalence.36,50
Biosimilars are developed using a method of ‘reverse engineering’ as the manufacturer does not have direct access to data for the reference product at any point and, as such, a manufacturer must thoroughly analyse reference products available on the market, as well as utilise the scientific literature and general knowledge to understand how to develop cell lines and associated processes to manufacture a highly similar biological product.50 By continually refining their manufacturing processes, biosimilar manufacturers will eventually produce a molecule that is as close to the reference product as possible.50 Once this has been achieved, the key focus becomes characterising the biosimilar and comparing it with the reference product to provide data to support an application for marketing approval where the regulatory authorities will look to determine the overall clinical effect of the biosimilar versus the reference product.50 The documentation supporting a request for marketing approval for a biosimilar encompasses extensive comparative detail on the physiochemical and biological properties of the biosimilar and its reference product, as well as preclinical, pharmacokinetic, and bioequivalence data. A limited amount of clinical data obtained using a sensitive endpoint in at least one indication approved for the reference product is needed to confirm no differences exist in efficacy and safety compared with the reference product.50 Once a biosimilar has established its comparability with its reference product in at least one sensitive indication, and provided that it is scientifically justified, the biosimilar’s indications can be extrapolated to all indications approved for the reference product.50
The regulatory concept of proving comparability is not new and is fundamental for all biologicals, as the manufacturing process for reference products can be expected to change over time as part of a normal product ‘life cycle’.36 For example, >35 changes have been made to the manufacturing process for infliximab since marketing approval was granted in 1999.36 Even minor changes in manufacturing must undergo regulatory review, whereby the characteristics of the product pre and post-change are compared. To facilitate this, both the EMA51 and FDA52 have developed guidelines for demonstrating comparability following a change in manufacturing process.
These comparability guidelines are issued by the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) and Committee for Medicinal Products for Human Use (CHMP) and set the data requirements for demonstrating comparability pre and post-manufacturing change for biological products, taking into account the magnitude of the change and its potential impact on the product.51,52 This process has been labelled the ‘comparability exercise’, and relies on comparing physicochemical testing, biological assays, and in some cases, non-clinical and clinical data.36
The ICH stipulates that comparability does not mean that these attributes are identical pre and post-change, but that they are “highly similar” and that any differences will have no impact on product efficacy or safety.51 For example, following a manufacturing change to the rituximab reference product, which resulted in changes in glycosylation and a subsequent increase in antibody-dependent cellular toxicity potency, on assessment, regulators concluded that the benefit-to-risk profile of the product was unchanged based on the totality of evidence available through pre and post-change characterisation, and no further clinical data were required.53
The same scientific principles apply to the generation of a biosimilar. One of the key questions from regulators is what clinical data is needed to show comparability and provide evidence to support an extrapolated indication. A regulatory authority will review the totality of evidence gained from the comparability exercise, and assess whether safety is clearly demonstrated, an increase in immunogenicity has been excluded, and if there are any remaining uncertainties, and then identify where further data is needed. The regulatory authorities are guided by several key principles when assessing the scientific justification for an extrapolated indication, including:
- Clinical experience with the reference product
- Established comparability
- Any differences in mechanism of action or safety (including immunogenicity) between indications
- Target receptors
An extrapolated indication is only likely to be granted for a similar or less sensitive indication or population, and this decision must be based on available clinical data (Figure 3).26
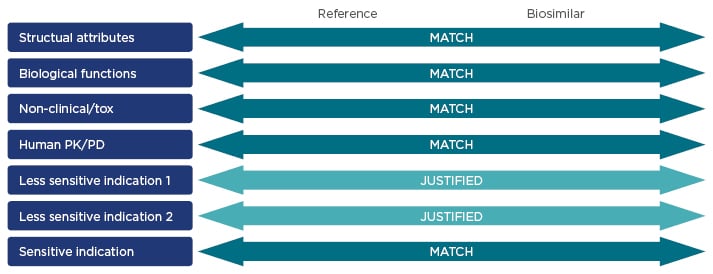
Figure 3: Key characteristics of reference products and biosimilars that are assessed during the regulatory approval process.16,26,54
PK: pharmacokinetics; PD: pharmacodynamics; tox: toxicity.
Therefore, extrapolating an indication is not a new process,26 and regulatory authorities have developed rigorous and scientific guidelines for assessing the comparability of therapeutic molecules that are applicable across various scenarios from a change in manufacturing process to the generation of a biosimilar.20,34,51,52 Extrapolation involves an extensive comparability exercise and regulatory authorities follow a key set of principles to establish stringent scientific justification before an extrapolated indication will be approved.26