
Abstract
Inherited thrombocytopenias comprise a heterogeneous group of blood disorders with abnormalities in genes related to glycoproteins and adhesion molecules, signalling pathways, cytoskeletal components, granule formation, and transcription factor complexes. Recent improvements in sequencing technology have increased the number of transcription factor mutations that have been implicated as causative for these platelet disorders. Mutations in RUNX1, GATA1, GFI1B, FLI1, and ETV6 share common features, including a variable bleeding history often associated with abnormal but non-specific changes in platelet morphology and platelet function testing. The phenotype of the underlying platelet disorder is often variable despite mutations in the same transcription factor, suggesting that the site of mutation and the protein domain that is perturbed is an important determinant of the clinical syndrome. Importantly, some of these transcription factor mutations are associated with other physical abnormalities, including an increased risk of acute leukaemia as well as solid organ malignancies. Genetic diagnosis of these disorders allows rational medical management to prevent bleeding, as well as providing an opportunity for family screening in order to reduce disease burden.
INTRODUCTION
Platelets mediate primary haemostasis after tissue trauma. Abnormalities in the number or function of platelets may predispose to spontaneous mucosal bleeding, or may precipitate problematic bleeding at the time of surgery. The correct diagnosis of platelet disorders provides an opportunity to prevent bleeding episodes and allows optimisation of haemostatic function for surgery. Isolated thrombocytopenia is a relatively common abnormality detected on full blood count analysis with most cases resulting from an increase in consumption of platelets in the circulation or a failure of adequate production from the bone marrow. Common causes include autoimmune or drug-induced consumption, sepsis, immunodeficiency syndromes, or bone marrow suppression from malignancy. Inherited causes of thrombocytopenia are caused by a range of genetic defects and are important to correctly identify; isolated thrombocytopenia has been misdiagnosed as immune thrombocytopenia in a number of clinical cohort studies with subsequent inappropriate treatment using corticosteroid therapy and splenectomy.1,2 Traditional laboratory investigation of inherited causes of thrombocytopenia includes assessment of blood film morphology, functional aggregation studies, and flow cytometry.3 In recent years, an increasing use of genetic technologies with DNA sequencing has allowed better molecular characterisation of platelet disorders and provided a platform for the discovery of new genetic defects that are causative of platelet-related bleeding.
THE ROLE OF TRANSCRIPTION FACTORS IN MEGAKARYOPOIESIS AND PLATELET PRODUCTION
Megakaryopoiesis and platelet production occur through a series of co-ordinated differentiation and maturation events that are orchestrated by cytokines and tightly regulated by networks of haematopoietic transcription factors.4,5 Different transcription factors have been identified to act at various stages of haematopoiesis,4 and in the megakaryocyte lineage a complex of key transcription factors (GATA2, LYL1, TAL1, ERG, FLI1, RUNX1, LMO2) expressed by haematopoietic stem cells is considered to initiate the development programme.5 These ‘prime’ sets of lineage-specific genes4,5 such as GP1BA and GP1BB, are uniquely expressed in platelets and provide haemostatic potential. The principal components of this complex are the GATA-binding transcription factors (GATA1 and GATA2), E twenty-six (ETS) factors (FLI1 and GABP-α), and the transcription activator-like helix-loop-helix factors that regulate target genes and interact with other factors.4
Runt-Related Transcription Factor 1
The core binding factor (CBF) is a haematopoietic transcription complex that is composed of α (CBF-α) and β (CBF-β) subunits. RUNX1 is one of three possible α subunits (RUNX1, RUNX2, or RUNX3) that binds to DNA via a conserved 128-amino-acid runt homology domain (RHD). CBF-β does not bind DNA directly but binds the RHD and stabilises the complex.6 RUNX1 acts on numerous target genes and proteins that are important in megakaryocytic maturation (for example MYH10), and platelet formation (MPL, MYL9, MYH9, TUBB1/2) and function (protein kinase C-theta and integrin αIIbβ3).7-10
Germ-line mutation of RUNX1 on chromosome 21q22.12 is associated with an autosomal dominant disorder called familial platelet disorder with propensity to acute myeloid leukaemia (FPD/AML).11-13 FPD/AML is characterised by dysmegakaryopoiesis, thrombocytopenia with normal sized platelets (Figure 1), a platelet function defect, and a propensity to develop myelodysplasia or acute leukaemia.8,9,11-16 Mutations causing FPD/AML commonly affect the RHD and less commonly the C-terminal transactivation domain.17 Affected individuals have a variable mucosal bleeding tendency from childhood ranging from asymptomatic to moderate.17-19 Most individuals with FPD/AML have mild-to-moderate thrombocytopenia, however normal platelet counts have been documented in a number of family pedigrees and do not exclude the presence of a significant RUNX1 mutation.18,19 Similarly, the platelet functional defect detected by light transmission aggregometry may be variable and has included reduced responses to collagen, adenosine diphosphate (ADP), arachidonic acid (AA), and epinephrine (Table 1). These changes are attributed to a storage pool deficiency and in some cases resemble an ‘aspirin’ like defect.8,11-13,15,16,20 A recent review of 41 carriers with RUNX1 alterations in nine unrelated French families described platelet dense granule release defects in 10 patients, regardless of the type of RUNX1 mutation, suggesting a link between this defect and FPD/AML.18 Further qualitative platelet dysfunction is caused by impaired integrin αIIbβ3 activation.8,20
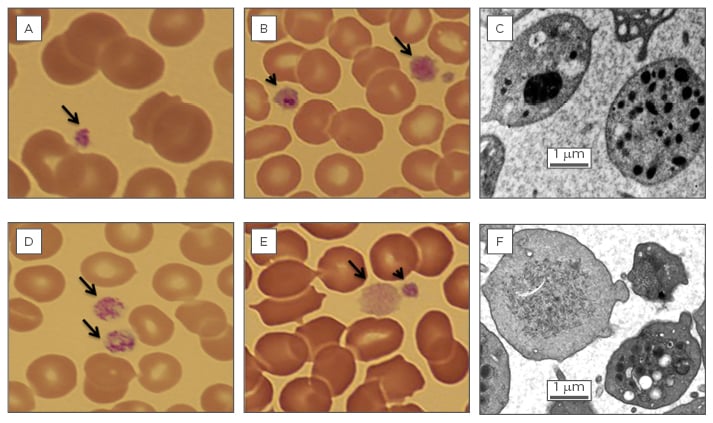
Figure 1: Platelet abnormalities associated with haematopoietic transcription factor mutation.
A) Photomicrograph of a blood film (100x) from an individual with a RUNX1 C-terminal transactivation domain mutation. A normal sized platelet with normal granulation is seen (arrow).
B) Photomicrograph of a blood film (100x) from an individual with the FLI1 R324W Two large abnormal platelets are seen (arrow and arrow head). One of these platelets (arrow head) appears to contain a giant granule.
C) Transmission electron micrograph from an individual with the FLI1 R324W mutation showing a platelet with a giant fused α-granule and a platelet with normal α-granule distribution.
D) Photomicrograph of a blood film (100x) from an individual with the GFI1B C168F Two large platelets with normal granulation are seen (arrows).
E) Photomicrograph of a blood film (100x) from an individual with the GFI1B H294fs A Large abnormal hypogranular platelet is present (arrow) as well as a normogranular platelet (arrow head).
F) Transmission electron micrograph from an individual with the GFI1B H294fs mutation showing both an agranular platelet and a platelet with normal α-granule content.
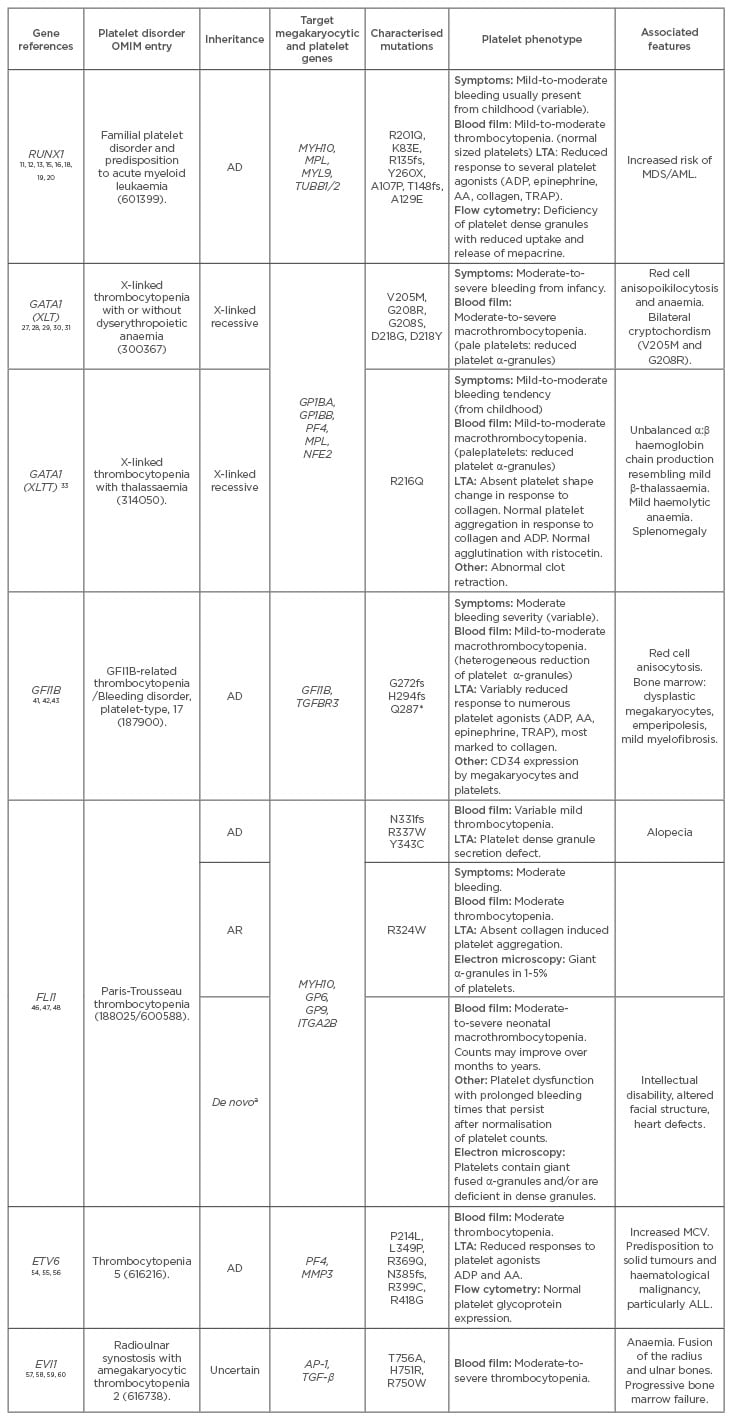
Table 1: Genetic and clinical features of transcription factor mutations.
AA: arachidonic acid; AD: autosomal dominant; ALL: acute lymphoblastic leukaemia; AR: autosomal recessive; LTA: light transmission aggregometry; MCV: mean cell volume; MDS: myelodysplastic syndrome; AML: acute myeloid leukaemia; OMIM: Online Mendelian Inheritance in Man; TRAP: thrombin activating peptide; ADP: adenosine diphosphate.
a Familial cases are rare with most individuals having de novo isolated 7-16 Mb deletions. Occasional individuals with AD inheritance have been reported.49
¶ Mutations of RUNX1 selected from OMIM. Over 27 mutations in 36 pedigrees have been identified.65
The evolution to leukaemia appears partly determined by the functional impact of the specific RUNX1 mutation.14,16 Mutations of either the RHD or C-terminal domain that alter the ability of RUNX1 to bind DNA but retain an ability to heterodimerise with CBF-β, generate a dominant negative phenotype that interferes with the function of the wild-type allele.21 These dominant negative acting mutations are associated with a higher risk of leukaemic transformation (over 40% in some cases) compared with mutations that induce haploinsufficiency by reducing levels of wild-type RUNX1 with resultant deregulation of key target genes, causing genomic instability and the potential to acquire additional somatic mutations, sometimes affecting the second RUNX1 allele.14,17 An ability to predict the phenotype based on the mechanism of action of the RUNX1 variant is therefore desirable and not possible without utilising diagnostic sequencing platforms.
Moreover, although the detection of a dense granule release defect in the presence of an autosomal dominant thrombocytopenia with normal platelet size may raise the suspicion of FPD/AML, the variable bleeding tendency, mild thrombocytopenia, and absence of morphological platelet abnormalities on the peripheral blood film (Figure 1) make the diagnosis of RUNX1 mutations (solely by phenotypic testing) very difficult without a high index of clinical suspicion based on family history. Optimal management of affected individuals and their families is largely based on expert opinion.22 Recommendations include: referral to a specialist team consisting of a physician and genetic counsellor, initial blood tests comprising full blood count and full human-leukocyte antigen typing of the patient and their first degree relatives (in the event that a bone marrow transplant is required in the future), bone marrow biopsy (to detect occult malignancy), and subsequent regular full blood count assessment and biannual haematological review.22
Globin Transcription Factor 1
The GATA1 transcription factor is encoded by GATA1 on chromosome Xp11.23 and is expressed in erythroid cells, megakaryocytes, mast cells, and eosinophils.23 During megakaryopoiesis, GATA1 assists in lineage commitment of ‘primed’ genes and, in concert with GATA2 and other transcription factors, including FLI1 and GABPA, regulates the activation of megakaryocyte and platelet specific genes such as GP1BA, GP1BB, PF4, MPL, and NFE2.4,24
GATA1 has two important functional domains, the N terminal domain that mediates transcriptional activation and the C terminal zinc finger domain. The zinc finger domain is required for an interaction with its transcriptional co-factor, the friend of GATA1 (FOG1), and also influences GATA1 binding affinity to DNA at specific genomic sites.25,26 The location of GATA1 mutations within these protein domains has been correlated to clinical phenotypes. To date, all known mutations resulting in GATA1-related cytopenias affect the amino terminal zinc finger domain of GATA1. Five variants (V205M, G208R, G208S, D218G, and D218Y) have been reported to affect the FOG1 binding face of the amino terminal zinc finger.27-31 These decrease the GATA1 to FOG1 interaction but preserve GATA1 to DNA binding ability and cause X-linked thrombocytopenia (XLT).25 The severity of the clinical phenotype is dependent on the degree of the GATA to FOG1 interaction disruption caused by the mutation.31 Individuals with V205M, G208R, and D218Y have the most severe phenotype characterised by mucocutaneous bleeding from infancy, macrothrombocytopenia (some platelets displaying reduced granulation on the blood film), red cell anisopoikilocytosis, and severe anaemia requiring transfusions (Table 1). Interestingly, two male half siblings and an unrelated male child with V205M and G208R, respectively, had cryptorchidism.27,28 A milder phenotype was described in men from two unrelated families carrying G208S and D218G mutations.29,30 In these cases, the phenotype was characterised by profound bleeding, macrothrombocytopenia, and mild dyserythropoiesis without anaemia. X-linked thrombocytopenia with thalassaemia (XLTT) is caused by mutations affecting the DNA binding face of the amino terminal zinc finger of GATA1 resulting in decreased GATA1 binding to DNA.32 A single variant, R216Q, has been reported in multiple families causing XLTT. This mutation causes milder anaemia and macrothrombocytopenia than those individuals with XLT.33 In addition, a distinguishing feature is unbalanced α:β globin chain synthesis with the production of abnormal α3:β1 and α4 haemoglobin molecules, creating a phenotype resembling mild β-thalassaemia.25,33 Notably, platelets in XLTT have reduced α granules, giving the appearance of grey platelets on the blood film (Table 1).33
Growth Factor-Independent 1B
Growth factor-independent 1B (GFI1B) is a member of the GFI zinc-finger transcriptional repressor family. Its expression in differentiated cells is restricted to erythroid and megakaryocytic cells where it plays an essential role in development.34,35 GFI1B contains an N-terminal repressor Snail/Gfi1 (SNAG) domain, which binds protein complexes that mediate transcriptional change, and six C-terminal zinc fingers that demonstrate sequence specific DNA binding.36 It is predicted that zinc fingers 3, 4, and 5 directly bind to DNA, while the functions of zinc fingers 1, 2, and 6 are not known.37 GFI1B regulates megakaryocytic genes controlled by other lineage-restricted regulators, such as GATA1, FLI1, SCL1, TEL/ETV6, RUNX1, MKL2, and SRF, and binds to its own promoter, auto-regulating its expression.38,39
Ten inherited mutations of GFI1B associated with thrombocytopenia have been documented.40-42 Seven patients with non-synonymous single-nucleotide polymorphisms were detected by analysis of the GFI1B locus in 529 exome-sequenced cases enrolled by the BRIDGE consortium.40 The best characterised mutations, G272fs, H294fs, and Q287*, disrupt the fifth DNA-binding zinc finger and produce a bleeding disorder with an autosomal dominant pattern of inheritance (Table 1).41-43 Mild bleeding symptoms were reported in the proband who carried the G272fs mutation.43 Individuals with the H294fs mutation experience variable bleeding; some experience significant bleeding including urological bleeding (requiring nephrectomy) and intracranial bleeding, while other members of the family report only bleeding following surgery.41 Laboratory features for both families include macrothrombocytopenia and platelets with heterogeneous granulation with some platelets appearing relatively agranular (Figure 1).41,43,44 Notably, a platelet function defect was demonstrated by light transmission aggregometry in the three individuals tested carrying the H294fs mutation. All three individuals had absent responses to collagen and variably reduced responses to platelet agonists ADP, AA, epinephrine, and thrombin receptor-activating peptide. Similarly, individuals with the Q287* mutation have moderate thrombocytopenia (range: 42–116×109/L) with moderate-to-severe bleeding symptomatology on bleeding scores.42 Platelets were agranular on blood film examination and pleomorphic megakaryocytes were found on bone marrow examination in association with mild myelofibrosis.42 Functionally, the G272fs, H294fs, and Q287* mutations de-repress transcription of GFI1B-related target genes and inhibit wild-type GFI1B transcriptional activity in a dominant-negative manner.41-43 These transcriptional changes cause deficiency of key haemostatic proteins with marked reductions in platelet fibrinogen and P-selectin, and an increase in CD34 expression.42,43 Preliminary data from a mutation predicted to disrupt the first non-DNA binding zinc finger of GFI1B demonstrate phenotypic variability for abnormalities in this transcription factor. The C168F mutation has been reported by two groups40,45 and alters the conformation of the first zinc finger but does not disrupt transcription to the same degree as mutations altering the fifth zinc finger. The affected individuals appear to have a mild clinical phenotype with moderate thrombocytopenia (range: 78–121×109/L) but no bleeding history, and platelets with normal granulation.45 It is likely these individuals would not be diagnosed with traditional diagnostic platelet studies, but correct classification may be important as their mild phenotype may predict for reduced medical intervention at the time of surgery.
Friend Leukaemia Integration 1
Friend leukaemia integration 1 (FLI1) is a member of the ETS family of transcription factors and is predominantly expressed in vascular and haematopoietic tissue.46 Point mutation and small deletion in the ETS DNA-binding domain of FLI1 are reported as causative of a bleeding disorder with abnormal platelet function (Table 1). Three families with autosomal dominant inheritance of a bleeding disorder associated with FLI1 mutation were identified in the UK GAPP study.46 These individuals had a mild bleeding disorder with evidence of defective secretion of dense granules on laboratory testing.47 A further two siblings were reported in an Australian cohort48 that had a bleeding disorder with a recessive pattern of inheritance. These two siblings had moderate mucosal bleeding symptomatology, moderate macrothrombocytopenia, and prominent abnormalities in collagen induced platelet aggregation (Figure 1) (Table 1). All reported point mutations appear to perturb transcription at the recognised FLI1 target GP6 gene with subtle differences in phenotype, probably reflecting changes in the site of the described mutations in the ETS DNA-binding domain. Protein modelling predicts the autosomal dominant mutations alter the third α-helix that directly binds DNA, whereas the recessive mutation, R324W, is positioned outside the third α-helix and may alter the protein conformation of FLI1.48
Hemizygous deletion of the entire FLI1 locus is also suggested to be responsible for the platelet defect associated with Jacobsen syndrome. Jacobsen syndrome is a multisystem disease characterised by developmental delay, short stature, congenital heart disease, and dysmorphic facial features.48 It is due to a variably-sized deletion of the terminal end of chromosome 11q. Approximately 90% of individuals with Jacobsen syndrome have a platelet-related bleeding disorder called Paris-Trousseau thrombocytopenia.49 Patients with Paris-Trousseau thrombocytopenia typically have neonatal thrombocytopenia that may improve with age, however platelet function remains abnormal throughout life. Paris-Trousseau platelets are variable in size and 15% of circulating platelets contain abnormal, large fused α-granules that are characteristic of this disorder.50 Various genes in the deleted region have been proposed to be causative of the various organ pathologies with three lines of evidence, implicating FLI1 as responsible for the platelet defect. Firstly, small terminal deletions of 11q, not involving the FLI1 locus, are rarely reported to be associated with a partial Jacobsen syndrome and these individuals do not exhibit platelet pathology.51 In vitro studies of megakaryocytes derived from individuals with Paris-Trousseau thrombocytopenia suggest overexpression of FLI1 may rescue some features of the defect52 and finally, the characteristic giant fused α-granules present in Paris-Trousseau thrombocytopenia have also been demonstrated in the two individuals with the R324W mutation of the DNA-binding domain of FLI1 (Figure 1).48
A mild bleeding disorder with thrombocytopenia and decreased α-granules is caused by decreased binding of FLI1 and RUNX1 to the 5’ untranslated region of their transcriptional target gene, ANKRD26. This transcription factor binding defect leads to a failure of normal transcriptional silencing of the transcript during megakaryocyte development with subsequent abnormal cytokine signalling, causing thrombocytopenia and an observed predisposition to leukaemia.53
ETS Variant 6
The transcriptional repressor, ETS variant 6 (ETV6), is also a member of the ETS family of transcription factors. Somatic mutations in ETV6 are recognised in haematological malignancy and deletion of the transcription factor in mice causes failure of haematopoiesis within the bone marrow.54 Individuals with point mutations and small deletions in the ETS DNA-binding domain and the adjacent central domain of ETV6 have moderate thrombocytopenia with normal platelet volume that is inherited in an autosomal dominant manner.55-57 Some of these family members have bleeding symptomatology and at least one family had features of altered α-granule production with morphological evidence of granule elongation.55-57 Other haematological parameters are unaffected with the exception of an elevated mean corpuscular volume reported in some families.55 A non-specific platelet functional defect with reduced responses to agonists ADP and AA was seen in one family harbouring a P214L mutation, but was not observed in another family with the R418G mutation (Table 1).55 These described mutations act in a dominant-negative fashion to decrease transcriptional repression at the promoters of target genes PF4 and MMP3 as measured by luciferase reporter assays.55 The described mutant ETV6 proteins also appear to dimerise with wild-type protein and promote aberrant cellular localisation of the transcription factor to the cytoplasm. Importantly, individuals in many of these families demonstrate an increased incidence of haematological malignancies and solid tumours, including colorectal cancer. There was a marked predominance of acute lymphoblastic leukaemia described with 10 cases reported in the eight unrelated families identified with germline ETV6 mutation.55-57
Ecotropic Viral Integration Site 1
The MECOM gene encodes EVI1 and MDS-EVI1 proteins from different transcription start sites. EVI1 is a transcriptional regulator with multiple zinc fingers that is normally expressed in haemopoietic tissue and is associated with a poor prognosis when over-expressed or mutated in myeloid leukaemia. Three distinct missense mutations predicted to disrupt the DNA binding function of the 8th zinc finger domain of EVI1 have been described in three unrelated children.58 These children were born with moderate-to-severe thrombocytopenia associated with fusion of the radius and ulnar. Two of the children displayed concomitant anaemia and developed progressive bone marrow failure that required haematopoietic stem cell transplantation. Deletion of the entire MECOM locus and mutations in the homeodomain transcription factor, HOXA11, have been associated with very similar phenotypes.59-61
DIAGNOSTIC STRATEGIES
Diagnosis of inherited thrombocytopenia and platelet function disorders are hindered by lack of consensus regarding a standardised approach to testing, as well as diagnostic criteria for the conditions themselves.62 This has prompted international groups and societies to draft guidelines63,64 suggesting a logical step-wise approach using clinical symptoms and signs together with combinations of predominantly phenotypic screening and discriminatory assays to diagnose these conditions.62 Genetic testing in these algorithms is generally recommended as a third-tier investigation.64 Transcription factors interact with each other and multiple target genes in a combinatorial manner,4,5 therefore mutation of a single transcription factor may disrupt the expression of multiple genes that are important in megakaryocyte maturation. The result, in many cases, is a complex platelet phenotype that is further influenced by the site of the mutation. Hence, it is not surprising that, despite multiple phenotypic tests that interrogate only isolated structural elements and/or functional pathways in platelets, a diagnosis is not achieved. An argument could be made that DNA-based diagnosis be prioritised in the diagnostic algorithm aiming for a ‘one-stop one-step’ diagnosis for patients. Sequencing in this context may provide a definitive diagnosis as well as indicate the likely impact of the mutation, based on the location of the mutation within a specific protein domain. Accurate diagnosis of these disorders may avoid the misdiagnosis of immune thrombocytopenia as well as allowing other members of the family to be screened to minimise bleeding events and identify inherited risk of oncogenesis.






