
Abstract
The actin cytoskeleton plays many important roles in the lifecycle of platelets, from biogenesis from megakaryocytes, to activation and clearance from the circulation. It is therefore unsurprising that mutations in genes regulating the dynamics of this cytoskeleton lead to numerous inherited thrombocytopenias. A diverse array of proteins are affected, including actin nucleators, structural proteins, myosin motors, and transcriptional regulators. This review summarises the current understanding of how genetic dysregulation of the actin cytoskeleton can contribute to the pathogenesis of thrombocytopenia.
INTRODUCTION
Thrombocytopenia is diagnosed when a patient’s platelet levels fall below the normal range of 150,000–450,000 platelets/µL of blood leading to defective blood clotting. A low platelet number may be caused by decreased platelet production, increased destruction, or sequestration from the circulating blood.1 This can be aggravated if the platelet function is further impaired. Many of these activities rely on the flexible and rapidly responsive nature of the actin cytoskeleton (AC), where filamentous actin (F-actin) can be rapidly assembled and disassembled from globular actin (G actin) monomers. This dynamic is regulated by a number of interacting molecules with various functions. These include those which potentiate the nucleation or branching of actin filaments, those which bind monomeric actin sequestering it from the F/G-actin equilibrium, and those which bind filaments cross-linking and stabilising, or capping and inhibiting further polymerisation. Furthermore, myosin motors can move along actin filaments, shuttling ‘cargo’ or generating contractile force. The importance of these mechanisms is discussed within this review in the context of genetic mutations which affect these activities leading to thrombocytopenia (Table 1).
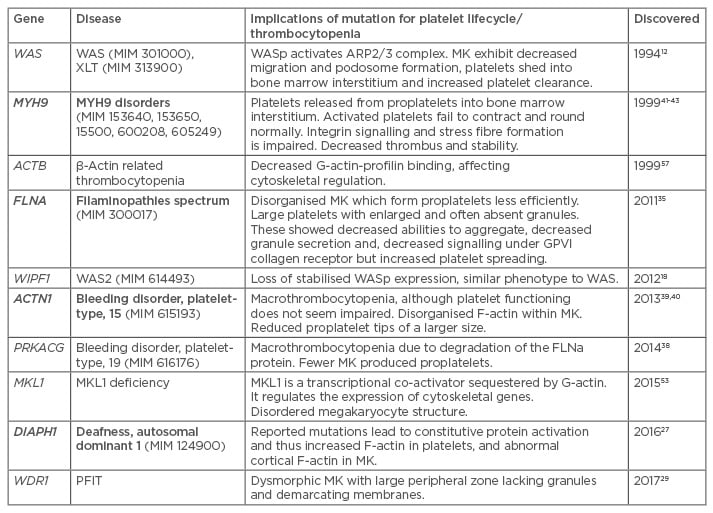
Table 1: Summary of AC-regulating genes associated with thrombocytopenia.58
Bold indicates dominant inheritance.
MK: megakaryocyte; G-actin: globular actin monomers; F-actin: filamentous actin; WAS: Wiskott–Aldrich syndrome.
PLATELET LIFECYCLE
Platelets are produced by megakaryocytes (MK) in bone marrow and in the lung,2 a process which is highly dependent on both the actin and tubulin cytoskeletal systems.3,4 MK develop from haematopoietic stem cells through endomitosis, giving them polyploid nuclei and an accumulation of cytoplasm. This facilitates the mass production of platelet components, including the generation of α and dense granules. MK have an extensive internal demarcation membrane which supplies an outer-membrane for the platelets. These form from tubular invaginations of the cell membrane organised by the AC, and then supplemented with golgi vesicular membranes.5 Within the bone marrow, mature MK migrate to the blood vessels of the basement membrane, an actin mediated process, and degrade the basement membrane with actin-rich podosomes; this permits the extrusion of proplatelet processes into the sinusoids. Whilst the extrusion of the proplatelets is dependent upon microtubules, their bifurcation and, hence, increase in platelet producing area is dependent upon actin dynamics. The tip of these proplatelets then bud off as preplatelets under actin contractile forces. See Italiano3 and Poulter and Thomas4 for comprehensive reviews.
The structure of the platelets themselves is dependent on the actin and tubulin cytoskeletons. Actin is the most abundant protein in the platelets (constituting 20%) and forms an internal network of ~2,000 actin filaments, cross-linked by filamin A (FLNa).3 FLNa is another extremely abundant protein in platelets, and links actin filaments to surface receptors, such as the von Willebrand factor receptor, the Glycoprotein Ib/IX/V complex, and αIIbβ3. The activation of these receptors leads to intracellular signalling and cytoskeletal reorganisation, enabling platelet aggregation, contraction, and degranulation.6,7 Unactivated platelets have a lifespan of ~10 days in the systemic circulation. As platelets age, they become desialyated and are recognised by the Ashwell–Morell receptor and cleared in the liver.8 In immune thrombocytopenia, autoantibodies to platelets also trigger platelet clearance in the spleen and by stimulating desialyation (Figure 1).9-11
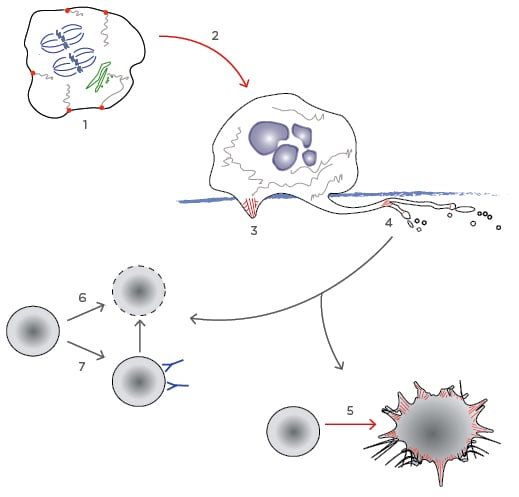
Figure 1: The platelet lifecycle.
Processes particularly dependent on the AC are highlighted in red.
1) The AC is involved in organising intracellular structure in promegakaryocytes. Endomitosis accumulates genetic material (blue) and formation of the actomyosin ring leading to cytokinesis is inhibited. PACSIN2 nucleates the formation of demarcation membrane system from the cell membrane and the golgi (green); 2) Megakaryocytes migrate to the basement membrane. Here they produce actin rich podosomes to degrade it, allowing; 3) the extension of proplatelet processes into the bone marrow circulation; 4) The AC stimulates branching of the proplatelet processes, and also the budding of preplatelets, which mature to become platelets; 5) When a mature platelet becomes activated, reorganisation of the AC leads to spreading and the extension of lamellipodia and filopodia precipitating clot formation; 6) As un-activated platelets age, they become desialyated and are cleared by Ashwell–Morell receptors in the liver; 7) In ITP, antiplatelet antibodies can trigger desialyation or stimulate clearance in the spleen.
AC: actin cytoskeleton.
DEFECTIVE FILAMENT BRANCHING IN WISKOTT–ALDRICH SYNDROME
Wiskott–Aldrich syndrome (WAS) is characterised by microthrombocytopenia (low counts of small platelets) recurring bloody diarrhoea, eczema, immunodeficiency, and often autoimmune disease. It is caused by recessive mutations in the WAS gene on the X-chromosome and is thus more prevalent in males.12 X-linked thrombocytopenia is caused by mutations in the same gene but manifests as microthrombocytopenia alone.13 The encoded protein, WASp, is restricted to the haematopoietic cell lineage. WASp integrates intracellular signalling with activation of the ARP2/3 complex; this complex then initiates the branching of new actin filaments from existing filaments, reviewed in Moulding et al.14 WASp is stabilised by WASp interacting protein (WIP); loss of WIP by mutation in the WIPF1 gene leads to absent WASp and a WAS disease phenotype.15 The loss of WASp function has multiple consequences in the platelet lifecycle, resulting in decreased platelet production and increased clearance.16 Whilst WASp-deficient MK can produce proplatelets and platelets normally in vitro,17 in vivo defects in the platelet production highlight the role of the AC in mediating the interaction between the MK and the cell microenvironment.18 MKs from WAS patients do not migrate efficiently to the bone marrow sinusoids and they do not extrude the actin podosomes to break through the basement membrane, and in fact release platelets aberrantly into the bone marrow interstitium.19,20 Platelets that do make it into the blood circulation are cleared more rapidly. This is proposed to be due to increased phagocytosis or antibody-mediated clearance.21-24 This can be mitigated in some patients or mice through splenectomy, although thrombocytopenia can reoccur if antiplatelet antibodies develop.25,26
CONSTITUTIVE DIAPH1 ACTIVATION: DEAFNESS, AUTOSOMAL DOMINANT 1
Formins are proteins which potentiate actin polymerisation from the barbed end of the filament, as well as regulate the microtubule cytoskeleton. DIAPH1 encodes the Rho-effector diaphanous-related formin 1. This protein is 1,272 amino acids long; heterozygous truncating mutations near the C terminus of this protein at amino acids 1,210 and 1,213 have been reported to lead to hearing loss and macrothrombocytopenia. These mutations result in the loss of the regulatory diaphanous autoregulatory domain (DAD) and hence constitutive activation.27,28 Other mutations in this gene have been associated with hearing loss but not necessarily thrombocytopenia. Cultured patient MK had abnormal cortical F-actin and platelet count had also increased levels of F-actin, indicating that late truncation does indeed have consequences for actin turnover; however, there were also demonstrable effects on microtubule turnover, and it is likely that both contribute to the full phenotype.27
PERIODIC FEVERS, IMMUNODEFICIENCY, AND THROMBOCYTOPENIA
WDR1 is a protein that facilitates cofilin mediated disassembly of actin filaments. Two siblings with a recessive missense WDR1 mutation have been reported to have thrombocytopenia in conjunction with immunodeficiency and autoinflammatory disease. It is probable that the thrombocytopenia represents defective platelet biogenesis, MK are small and often lacking a nucleus or demarcating membranes. They have enlarged peripheral zones lacking in granules and organelles.29 Mice with hypomorphic mutations in WDR1 exhibit macrothrombocytopenia and the same MK phenotype. There were increased MKs in both the bone marrow and especially in the spleen. Granules were abnormally distributed within platelets and the microtubules coils were irregular. The authors demonstrated, however, that clearance of these abnormal platelets was at the normal rate, and there was no autoimmune involvement dependent on B or T cells, and no splenic sequestration, demonstrating that decreased production was the primary cause of thrombocytopenia.30
THE FILAMINOPATHIES
FLNa is a fundamental component of the AC in most cell types, therefore mutation of this gene has severe consequences in development throughout multiple tissues.31 The FLNA gene occurs on the X-chromosome; therefore, mutations are usually heterozygous in females and lethal in males. Mutation results in a spectrum of diseases, including periventricular nodular heterotopia32 and otopalatodigital syndromes.33 Thrombocytopenia is often described in these patients,34,35 and furthermore, the FLNA mutation has recently been linked to a macrothrombocytopenia in the absence of congenital comorbidities.35,36 Platelets were of a large size, with enlarged and often absent granules. They showed a decreased ability to aggregate, decreased dense granule secretion, and decreased platelet signalling under glycoprotein VI (GPVI) collagen receptor, but not for G-protein coupled receptors. Platelets showed an increased propensity for spreading, suggesting increased ability of the AC to reorganise without restriction from crosslinking FLNa.35
A murine model with FLNa knockdown restricted to MKs showed that those MK release enlarged unstable platelets.37 X-inactivation in female patients generates platelet production from either wild-type (WT) MK or from those possessing mutation. Platelets lacking WT FLNa represent a minority of those circulating in the blood but it is unclear if this represents a decrease in production or an increased clearance. Whilst the MK compartment was expanded in the bone marrow (as also observed in the murine MK model), MKs lacking WT FLNa formed proplatelets less efficiently, and showed abnormal intracellular organisation.26,28 Soluble FLNa regulates the formation of the membrane invaginations that initiate the generation of the demarcation membranes,5 FlnA-null MK have defective demarcation membrane organisation.35,37
The regulation of FLNa levels is important in platelet production. PRKACG is a gene which encodes the γ isoform of a subunit of a cAMP-dependant protein kinase which phosphorylates FLNa. This protects FLNa from proteolysis. Homozygous missense mutations in this gene lead to macrothrombocytopenia and increased bleeding scores in two siblings. FLNa levels were depleted in mature MK cultured from patient CD34+ cells. The MK differentiated normally, but a smaller proportion had formed proplatelets.38
α-ACTININ-1 RELATED BLEEDING DISORDER
Another protein which crosslinks actin filaments in bundles is α-actinin-1. A number of dominant missense mutations have been linked to macrothrombocytopenia and anisotropy. Platelets seemed to function normally in spreading, aggregation, and clot retraction.39,40 However, MK appeared abnormal with reduced numbers of proplatelet tips which were of a larger size, perhaps indicating a defect in their bifurcation.40 Patient MK, as well as murine MK, transduced with a mutant protein, exhibited disorganised F-actin filaments.39,40
THE MYH9 DISORDERS
Mutations in MHY9 are linked to a spectrum of syndromes with macrothrombocytopenia (May–Hegglin anomaly, Fechtner syndrome, Sebastian syndrome, and Epstein syndrome).41-43 Universal symptoms are thrombocytopenia, large platelets, and leukocyte inclusions composed of ribosomes, endoplasmic reticulum, and abnormal myosin aggregates.44 Other symptoms can include cataracts, nephritis, and sensorineural hearing loss.41-43 The disease phenotype is influenced by the position of the mutation within the encoded protein non-muscle myosin heavy chain IIA. Non-muscle myosin II (NMM-II) is a complex of two heavy chains and four light chains. The N-terminal of the heavy chain binds to actin filaments revealing the adenosine triphosphate binding site. Myosin light chains bind the neck region of the heavy chains, and when the light chains are phosphorylated, the complex is activated and can bind actin filaments and initiate the contractile response. The C-terminal assists actin polymerisation and cargo-loading; reviewed in Althaus and Greinacher45 and Balduini et al.46 Heterozygous mutations have a dominant negative affect because the WT protein becomes sequestered in intracellular aggregations with mutant protein.47,48
MK numbers in the marrow may be slightly elevated. They appear to develop normally, but, similar to WAS, platelets are released into the bone marrow interstitium rather than in the sinusoids. This is likely to be due in part to a defective Rho-associated kinase phosphorylation of the myosin light chains.49 Platelets aggregate and secrete granules normally, but they fail to contract. They can extend lamellipodia, although without stress fibres integrin signalling is impaired, which hinders thrombus growth and stability.50
MLK1 DEFICIENCY
The megakaryoblastic leukaemia (translocation) 1 (MKL1) gene encodes a myocardin-related transcription factor cofactor of serum response factor (SRF). It is sequestered by G-actin monomers and released upon polymerisation to F-actin, where it translocates to the nucleus, binds SRF, and induces the expression of a number of regulatory genes and β-actin itself.51,52 This includes MYL9, another component of NMM-II that associates with MYH9 encoded heavy chain.46 A female patient presented with immunodeficiency and mild thrombocytopenia, caused by homozygous nonsense mutation in MKL1.53 Whilst the patient’s MK were not studied directly, their neutrophils showed impaired migration and podosome formation was impaired in monocyte derived dendritic cells. It is possible that MK would acquire similar impairments. Knockdown of MLK1 expression in human megakaryocyte progenitors produced MK with disordered demarcation membranes and α granules. Migration was impaired with decreased formation of proplatelets, filopodia, lamellipodia, and stress fibres.54 MKL1 knockout mice exhibit thrombocytopenia and defective megakaryopoiesis.55,56 Record et al.53 hypothesised that the thrombocytopenia in MKL1 deficiency may have been relatively mild because MKL2 may be able to partially compensate for lost MKL1 activity, as observed in mice.56
β-ACTIN RELATED THROMBOCYTOPENIA
Intriguingly, a missense mutation in the β-actin protein itself has been linked to thrombocytopenia, immunodeficiency, and mental retardation.57 The glutamic acid residue at position 364 is substituted with lysine (E364K). Given the fundamental importance of this protein, it is perhaps unsurprising that this mutation did not effect in vitro actin polymerisation itself but rather may influence cytoskeletal regulation by affecting the binding of regulatory proteins. The authors demonstrated a clear negative effect of this mutation on profilin binding to G-actin; this depletes actin monomers from equilibrium with F-actin. They did not study platelet biogenesis or function directly, other than to note that the bone marrow MK population was increased, perhaps to compensate for depleted circulating platelets. They did demonstrate impaired migratory capacity in other cell types.57
CONCLUDING REMARKS
Numerous factors control the regulation of the dynamic AC throughout the platelet lifecycle. This is reflected in the number of actin-related genes which when mutated give rise to clinical thrombocytopenias. As we understand more about the molecular mechanisms controlling the AC and the pathogenesis of the thrombocytopenias, we are bound to continue to expand this list over the next few years. The capabilities of next-generation sequencing technologies have already facilitated the identification of several of the actin-related genes discussed above, as they permit the screening of large volumes of patients’ genetic material at once, including entire exomes and genomes. Another promising avenue is the development of multiplex sequencing panels, which can be designed to screen multiple known genes at lower costs. This will widen access to molecular screening for many patients, and may lead us to redefine and broaden the described phenotypes as there will be increased chances of identifying mutations in known thrombocytopenia genes manifesting with unusual presentations.





