
Abstract
The next-generation sequencing era has repeatedly demonstrated that the amount of acquired somatic mutations in paediatric cancers can rarely account for the total incidence of any cancer subtype. In addition, many cancer-related mutations can be found in healthy individuals. These findings strongly suggest that additional genetic or epigenetic variation is required for malignant transformation, particularly in children who have significantly less environmental exposure and resulting genetic damage. Current studies now suggest that 3–33% of paediatric cancer patients have a predisposition to cancer. These germline genetic or epigenetic changes are frequently found in molecular mechanisms regulating normal human development which have long informed our understanding of developmental biology. Blockade of development is a mechanism of transformation consistent with the higher number of immature cancer cell types in paediatric patients. Thus, while nearly every cancer is a combination of germline variation and somatic mutation, the relative contribution to tumourigenesis in paediatrics is weighted toward germline changes. This review will explore how paediatric predisposition to leukaemia is influenced by germline genetic and epigenetic variability of variable penetrance. Improved understanding of these critical developmental mechanisms will lead to improved surveillance and perhaps guide a new class of therapeutics aimed at promoting normal differentiation rather than widespread cytotoxicity.
INTRODUCTION
The next-generation sequencing era has facilitated exciting progress in understanding the varied aetiologies of cancer on a genomic level. This success has primarily come through a model of tumour/normal sequencing from the same individual, where the normal sequence is simply used to subtract germline variation from cancer and identify cancer-specific mutations. In children, however, these cancer-specific mutations are much less common,1 and very rarely does one find the 2–8 deleterious mutations thought to be necessary to transform a healthy cell to cancer.2 In fact, the paediatric leukaemia with the highest mortality rate (>50%), infant leukaemia, has the fewest somatic mutations of any sequenced cancer with an average of 1.3 non-synonymous mutations per genome.3 With respect to somatic mutations, this makes perfect sense given the rarity of paediatric cancers, fewer cell divisions, and decades less of environmental exposures compared with adults. Unlike adult cancers, where 5–10% have an identifiable genetic predisposition4,5 and appear primarily driven by acquired somatic mutation, mutations are mainly in mechanisms regulating cellular maintenance, division, and DNA repair.2 In contrast, the factors contributing to paediatric tumourigenesis are more likely to affect genes regulating age-specific mechanisms of differentiation and cell fate determination.6-9
Most paediatric cancer sequencing studies have focussed on identifying somatic coding mutations in leukaemia. The inability to account for paediatric cancer solely via somatic mutation suggests that additional contributory information may lie in the non-coding or inherited genome or epigenome. In this regard, a Swedish epidemiology study analysing >10 million individuals examined the risk of acute lymphocytic leukaemia (ALL) in full, non- twin siblings of children with the disease, and found an increased odds ratio of 7.08,10 consistent with a familial contribution to paediatric ALL. The variable penetrance of familial leukaemias suggests that these variants do not act by dominant modes of inheritance. If we accept that malignant transformation requires 2–8 ‘mutations’,2 then children must possess a combination of 2–8 (dependent upon effect size) inherited or acquired, genetic or epigenetic alterations that facilitate transformation. This review will focus on the relative contributions of germline variation, epimutations, and acquired somatic mutation to the aetiology of paediatric leukaemia, where more is known about the roles of these three types of variability. In simplistic terms, we can break this model down into an age-dependent schematic (Figure 1) and explore the interplay between these groups of genetic and epigenetic variability.
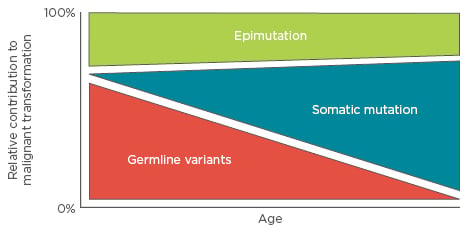
Figure 1: The relative contributions of germline variation, somatic mutation, and epimutation to malignant transformation as a function of age.
In this model, early childhood leukaemias depend on multigenic variability and mutation, and therefore may not show strong familial inheritance patterns as would be observed in a Mendelian disease.
GERMLINE VARIABILITY
If paediatric cancer were solely a disease of somatic mutation, the relative lack of somatic mutations in paediatric cancer would suggest the scant mutations found would have greater effect sizes, reducing the need for the additional mutations seen in adult cancers. Yet, when introduced into model systems and expressed at physiologic levels, the majority of these somatic changes fail to induce cancers that phenocopy the paediatric disease.11-13 This further suggests that somatic mutation alone is insufficient to explain the incidence of most paediatric malignancies, and that children’s cancer evolves by mechanisms distinct from adult cancer. Multiple studies suggest that anywhere from 3–33% of children with cancer have a predisposition toward malignancy,4,14,15 which presumably does not drive transformation but rather lowers the threshold for transformation. Indeed, many paediatric cancers appear to be derived from defects in normal developmental programmes,16 consistent with the majority of paediatric cancers arising from immature cell types (e.g. the ‘blastomas’, pre-cursor B cell ALL, germ cell tumours, etc.). This is in contrast to adults, who more commonly possess tumours derived from more terminally differentiated cell types (e.g. carcinomas) that have accumulated enough damage to become irresponsive to appropriate cell signalling.
Cancer Predisposition Syndromes
It has long been recognised that several genetic syndromes, which include a variety of pathognomonic clinical and phenotypic features, increase the risk of paediatric leukaemia and other cancers. Yet germline variation alone appears insufficient for malignant transformation. Individuals with Li–Fraumeni syndrome, BRCA1/2 variants, hereditary non-polyposis colorectal cancer, familial adenomatous polyposis, or other single cancer-related genetic syndromes typically do not develop cancer until the third decade of life, after enough cell divisions and the accumulation of additional genetic mutations.17,18 While individually rare, there are numerous paediatric cancer predisposition syndromes that are typically divided into functional classes: aneuploidy-associated predisposition, hereditary transcription factor syndromes, DNA instability syndromes, bone marrow failure syndromes, and differentiation defects. While each is briefly mentioned below, they have been reviewed in detail elsewhere.19
Aneuploidy-associated predisposition
One of the earliest recognised cancer predisposing conditions, Down’s syndrome (DS) is the archetypal aneuploidy-associated leukaemia. Children with DS have a 10 to 20-fold increase in leukaemia risk. The increase for acute megakaryocytic leukaemia (French-American-British classification M7) is even higher at a nearly 500-fold increased risk, such that when acute megakaryocytic leukaemia is seen in children without DS, there is often mosaicism or other anomalies in chromosome 21.20 There is a profound association to leukaemia in DS and somatic mutations in the X-linked gene, GATA1, generally in exons two or three, which leads to a clinically variable phenotype ranging from self-limited neonatal transient myeloproliferative disorder to fatal leukaemias in childhood. There is also growing evidence for cohesin mutations as leukaemia modulators,21 which are the topics of current investigation.
DNA instability syndromes
This group is well known due to the inability to repair double-stranded DNA breaks, leading to abundant somatic mutation. Well known instability syndromes are Li–Fraumeni and Bloom syndrome, ataxia-telangiectasia, and Fanconi anaemia, but there are other less common syndromes as well.17,19,22-25
Bone marrow failure syndromes
These are well-studied disorders that often result in myeloid, rather than lymphoid, leukaemia and consist of severe congenital neutropenia, dyskeratosis congenita, Diamond–Blackfan anaemia, thrombocytopenia and absent radii syndrome, Shwachman–Diamond syndrome, and congenital amegakaryocytic thrombocytopenia.17,19,22,26-28
Differentiation defects
With respect to leukaemia, juvenile myelomonocytic leukaemia is most commonly secondary to RAS mutations in neurofibromatosis Type 1 or PTPN11 mutations in Noonan syndrome,29,30 but a similar phenotype and acute myeloid leukaemia (AML) incidence has recently been described in individuals with germline CBL variants and developmental anomalies.31
Birth Defects
Within the past 10 years, there has been a growing body of epidemiologic literature recognising that children with birth defects (variably defined) have an increased risk of developing cancer. An examination of >3 million children in the Texas Birth Defect Registry found that children with birth defects harboured an incident rate ratio of 1.4 for developing leukaemia.32 While children with chromosomal birth defects have a greater risk of leukaemia (mainly due to DS), children with non-chromosomal birth defects are more likely to develop lymphomas, neuroblastoma, germ cell tumours, and central nervous system malignancies.33 Hypospadias, cleft lip, and hydrocephalus had no effect on cancer risk, but cleft palate, microcephaly, and renal or cardiac defects increased the risk of cancer nearly 3-fold.34 Recently, the AGORA database in the Netherlands has collected data on 3,747 children with birth defects, of whom 905 have childhood cancer. To date, the study has found that nearly 30% of these children develop ALL, the most common cancer in the registry, and another 17% have lymphoma.35 The fact that these defects in normal human differentiation carry an increased risk of childhood cancer further strengthens the link between abnormal development and cancer. Future studies should move beyond large-scale epidemiological surveys and focus on how the disrupted mechanisms resulting in congenital anomalies contribute to overgrowth and malignant transformation.
Inherited, Functional, and Single Gene Variants
In addition to syndromes and birth defects with clear phenotypic effects, there is growing evidence for cancer predisposition as a result of inherited germline variants in >60 different genes, without other signs or symptoms. As one would expect, this is the fastest growing category of genetic variation associated with paediatric leukaemia. Genome-wide association studies have identified heritable variation in IKZF1, ARID5B, CEBPE,36 and CDKN2A37 and larger regions of disequilibrium associated with paediatric de novo38,39 or ETV6-RUNX1-associated ALL.40
More recently, leukaemia predisposition (mainly to B cell ALL) has been associated with inherited single gene defects in transcription factors that skew normal haematopoietic differentiation For example, PAX5, required for normal B cell maturation, demonstrates heritable variants (mainly G547A) in multiple related individuals. These are transmitted in an autosomal dominant fashion with variable penetrance and predispose to lymphoblastic transformation due to B cell differentiation defects.41-43 ETV6 is another transcription factor required for nuclear localisation of transcriptional machinery, and is part of the most common translocation observed in paediatric leukaemia (ETV6-RUNX1). Exome and genome sequencing studies of paediatric pre-B cell ALL have identified hypomorphic germline variants that are defective in nuclear localisation, leading to aberrant target gene expression and a predisposition to B cell ALL that is typically accompanied by familial thrombocytopenia.44-46 More recently, PRDM9, important for regulating meiotic recombination events, was shown to be enriched for deleterious germline variants in multiple children with pre-B cell ALL.47 For AML, a condition of growing interestis familial platelet disorder with predisposition to AML secondary to hypomorphic germline RUNX1 variants that skew normal haematopoiesis in association with variable degrees ofthrombocytopenia and platelet dysfunction.14,19 However, germline RUNX1 variants have also been associated with ALL and thrombocytopenia48 and in patients with acquired mutations in ASXL1.49
Given that the germline variants appear to only establish a pre-leukaemic state, actual cases of leukaemia are almost certainly going to include a combination of multiple germline variants along with somatic mutations. For example, while infant leukaemia has the fewest non-synonymous point mutations per genome of any cancer sequenced to date, and over 75% of these cases harbour MLL-rearrangements, germline exomes from these children found a significant enrichment for rare, non-synonymous germline variants. In particular, bi-allelic variation in the pan-cancer tumour suppressor gene and histone methyltransferase, MLL3 (KMT2C), was a common feature.50,51 This pattern of inherited multi-allelic, multi-gene variation establishing a pre-cancerous state is likely to be increasingly recognised for many types of paediatric cancer that show a paucity of acquired mutation.
EPIMUTATIONS
With the realisation that inherited or acquired sequence changes cannot fully account for the incidence of paediatric cancer, the field has begun looking at other sources of variation. Research has shown epigenetic variability to be more diverse and complex than initially imagined. There is a qualitative difference in the nature of epimutation in paediatric versus adult cancer. Because many inherited variants or somatic mutations in developmental pathways interrupt gene regulation necessary for cell fate specification,52 there is a strong correlation between gene silencing as either the cause or the effect of other genetic events in paediatric cancer.16 In adults, somatic mutations are found in quite different epigenetic regulators (e.g. DNMT3A, IDH1/2 in adult AML) that are rarely variant or mutated in paediatrics. The true burden of epimutation as a function of age is not well described. The speculation is that there is probably a slightly bigger contribution from epimutation in paediatric leukaemia compared to adults due to the involvement of developmental mechanisms, which is depicted in Figure 1.
Secondary to Germline Variation or Somatic Mutation
Epimutations that occur secondary to germline variation or somatic mutation significantly overlap with the inherited transcription factor predisposition syndromes described above and have been reviewed for paediatric ALL elsewhere.53 Many of the most commonly described somatic mutations in paediatric leukaemias are found in epigenetic regulators: histone methyltransferases (MLL1/KMT2A, MLL2/KMT2B, EZH2), demethylases (TET2, KDM6A), polycomb repressor proteins (ASXL2) and histone acetyltransferases (CREBBP, EP300), and DNA methyltransferases and their regulators (DNMT3A, IDH1/2) among others.54,55 Epigenetic silencing of these genes may also occur due to environmental influences55,56 which may partially explain why the incidence of paediatric cancer has been steadily rising for the past 40 years.57 While the largest germline analysis to date of paediatric cancer patients finds the most common variant genes to be canonical autosomal dominant cancer drivers, TP53, APC, and BRCA2, the largest absolute number of variants were in the category including epigenetic modulators such as ARID5B, EP300, TET2, ATRX, and SETD2 among others, and 51% of these patients had leukaemia.15 Without appropriate gene expression during different embryonic or developmental stages, the barrier to malignant transformation is apparently lowered and uncontrolled proliferation results as the affected cells attempt to bypass whatever developmental blockade is encountered.
Inherited (Causal)
The actual heritability of epimutations is a topic of some debate as epigenetic signals are generally thought to be ‘erased’ after fertilisation and gradually re-established during embryogenesis and development.52,58 Mendelian inheritance of single nucleotide or copy number variants alter DNA methylation in a manner that inappropriately silences (tumour suppressors) or activates (oncogenes) gene expression as an initiating event in tumourigenesis.52,58 This mechanism was reported in a family with chronic lymphocytic leukaemia due to a rare variant promoting silencing of DAPK1.59 However, irrespective of an identified sequence change, the same DNA methylation-mediated silencing of MLH1 and MSH2 in all tissues from affected individuals in pedigrees with Lynch syndrome,60 suggesting that heritable epimutations may arise from multiple mechanisms and result in cancer predisposition. While many studies have evaluated DNA methylation in various paediatric leukaemias for diagnostic or prognostic value, the contribution of inherited epimutations in paediatric leukaemia is not well characterised.
ACQUIRED SOMATIC MUTATION
Paediatric cancer is marked by a high prevalence of canonical and cryptic translocations, but nearly all data point toward additional environmental, genetic, or epigenetic events having an additive effect leading to malignancy. In this model, we can envision a continuum where the risk of malignancy is dependent upon the abundance, the cumulative effect size of multiple genetic (germline and somatic), and epigenetic lesions. If this is the case, then these translocations should occur stochastically in the general population without resulting in malignancy due to the lack of the additive events. While many somatic mutations identified in cancer can occasionally be found in the germline of healthy individuals, prior searches to identify the true population-based incidence of translocations such as ETV6-RUNX161 and MLL-rearrangements62 have been highly controversial and no clear consensus on this topic exists. Alternatively, an aggregation of individually low penetrance variation where multiple variants are aggregated within a single individual and confer a greater additive effect size could account for the gap between the 8.5% of paediatric cancer patients with germline variation in known cancer genes.15 Additionally, 29% of paediatric cancer survivors (compared to only 5% of adult cancer survivors) meet criteria for predisposition when incorporating additional factors such as family history, cancer type, and comorbidities.4
CLINICAL APPLICATION
Longitudinal surveillance in sporadic and familial breast and colorectal cancer has markedly improved outcomes since implementation.63 Currently, no recommendations exist for systematic paediatric leukaemia screening, which is more likely due to the heterogeneity of the disease as opposed to the prevalence. When considering children with cancer predisposition, it is not always evident who should actually be screened or what combination of genetic (and epigenetic) lesions should be considered disease-related. For those that have clear phenotypic anomalies associated with a canonical syndrome or a strong family history, identifying at-risk individuals is straightforward even if the incidence of cancer is quite variable. In addition to who should be screened, an even more difficult question concerns which modality to use for screening. Genome sequencing, exome sequencing, methylation arrays, hybridisation- based methylomes, and bisulphite-treated genomes are all potentially informative. While the cost of genomic technology is gradually decreasing, these modalities are currently still too costly for governments, insurance companies, or individuals to offer for population-based screening. In addition, a majority of the data generated is often difficult, if not impossible, to interpret because we simply do not understand enough of the intricate interplay between these sources of genetic and epigenetic variability, giving these tests a high sensitivity, but a poor specificity.
In addition to screening, the invocation of congenital sequence or epigenetic variation that disrupts normal developmental mechanisms raises therapeutic concerns that are unique to paediatric cancer. While targeting developmental mechanisms may eliminate the leukaemia, there can be broad consequences to disrupting an essential developmental mechanism that often results in late-onset toxicities and long-term morbidities involving growth, cognition, fertility, and perhaps an even greater increased risk of secondary malignancies compared to patients without predisposition.64 An example of this is seen in murine models of Gorlin syndrome where inhibition of sonic hedgehog reduced rates of medulloblastoma but resulted in various defects in other systems.65 Thus, when it comes to predisposition toward paediatric leukaemia, even ‘targeted’ therapeutics may have untoward collateral damage if the primary goal of treatment is cytotoxicity.
However, if paediatric leukaemias are in fact the result of developmental blockades in normal haematopoiesis, perhaps an alternative strategy for leukaemia treatment can be extrapolated from acute promyelocytic leukaemia where the PML-RAR translocation arrests maturation of promyelocytes. The introduction of all-trans retinoic acid therapy allows the majority of promyelocytes to overcome this blockade and mature to myelocytes.66 This change in the acute promyelocytic leukaemia treatment approach, so-called ‘differentiation therapy’, has significantly reduced the need for cytotoxic therapy, resulting in improved outcomes. By identifying and overcoming blockades in normal haematopoietic development incurred in other paediatric leukaemias due to germline genetic or epigenetic variation, additional difficult-to-treat subtypes of paediatric leukaemia may enjoy improved outcomes with less long-term morbidity.
SUMMARY
Paediatric leukaemia is increasingly recognised to be the result of pre-existing predisposition. The landscape of germline variation, somatic mutation, and epimutation differs substantially from that seen in adults. The difference is not necessarily in the total amount of genetic or epigenetic damage inherent between the two groups, but in the source of that damage. Children are more likely to harbour germline variation that alters epigenetic regulation of developmental mechanisms, which are primarily active only in childhood. Identifying these mechanisms and their relative influence on paediatric leukaemogenesis will improve clinical screening protocols, which will hopefully occur in parallel with continued reductions in the cost of sequencing and a concomitant augmentation of our ability to accurately interpret the results. In addition, improved characterisation of these developmental pathways may inform novel therapeutics that depend less upon cytotoxicity and more on differentiation therapy to enable these malignancies to achieve their desired cell fate.





