Abstract
β-thalassaemia (BT) is a hereditary genetic blood disease caused by a mutation in the gene that encodes the haemoglobin protein. In the most severe forms, BT forces patients to undergo frequent blood transfusions, which has a significant impact on the quality of life. Classified as rare, BT is very common in the Mediterranean area, and is also found in the Middle East, Central Asia, India, South America, and North Africa.
This disease does not currently have a definitive cure, although technological progress and new gene therapies are achieving promising results.
This literature review was conducted with the aim to understand how BT affects patients’ lives in various social contexts in which they are involved. The authors also aimed to understand which methods are used for this assessment and the possible social actions that can help in the management of the disease.
Electronic databases, including PubMed, Scopus, and Web of Science, were used to search for the articles. Related article titles were selected and reduced to the abstracts of the relevant articles, after which the selected full articles were reviewed.
The reviewed articles showed consistent agreement in observing that the quality of life of patients with BT is considerably lower compared with the healthy population in terms of physical, emotional, social, and functioning at school. The negative results highlight the significance of the introduction of suitable programmes by healthcare providers, counsellors, and education authorities to provide psychosocial support, and improve academic performance. In addition, genetic counselling and intervention programmes would positively impact the lives of patients with thalassaemia.
My selection for this issue is the article ‘Social Impact and Quality of Life of Patients with β-Thalassemia: A Systematic Review’ by Greco and Marino. Quality of life is becoming more and more important in the evaluation of clinical trials, and also by regulatory authorities, particularly in chronic diseases (which can be seen by the U.S. Food and Drug Administration’s [FDA] recent position on luspatercept). Therefore, understanding its meaning and limitations is very important.
Key Points
1. β-thalassaemia (BT) is a chronic disease which affects patients’ lives in various social contexts as, in its most severe form, the condition involves blood transfusion dependence.2. The quality of life of people with BT is considerably poorer compared with the healthy population in terms of physical, emotional, and social aspects, as well as functioning at school.
3. It is essential to try to fill the gaps in the psychological support and social introduction of people with BT through a robust network of programmes, including family therapy, genetic counselling, educational interventions, and intervention programmes, emphasising the importance of spiritual growth, physical activity, and interpersonal relations.
INTRODUCTION
Thalassaemia is a group of hereditary microcytic haemolytic anaemias characterised by a defect in haemoglobin synthesis. β-thalassaemia (BT) is a hereditary disease transmitted by an autosomal recessive manner, characterised by deficiency (β+) or absence (β0) of synthesis of the β-globin chains of haemoglobin.1
The name thalassaemia derives from the Greek ‘thàlassa’ (sea) and ‘haîma’ (blood), and it was chosen due to the great distribution of this pathology in the Mediterranean area.1 BT is a condition of highly variable severity; it ranges from a form called thalassaemia minor, which is mostly asymptomatic, to the most severe form, known as thalassaemia major or Cooley’s disease, a condition that involves blood transfusion dependence (transfusion-dependent thalassaemia [TDT]).
Anaemia constitutes the predominant symptom, and transfusions can range from every 2–4 weeks to once every 2–3 months, depending upon the clinical severity of the disease. Repeated blood transfusion also prevents physical abnormalities associated with the bone marrow hyperactivity, which is responsible for the characteristic skeletal changes seen with thalassaemia.
Lifelong transfusions present several possible adverse effects, which include immunological reactions, development of antibodies to red cell antigens, transmission of infectious agents (hepatitis B and hepatitis C), and the accumulation of iron from the transfused red blood cells.2
From an epidemiological point of view, BT, together with other forms of thalassaemia, constitute one of the most widespread genetic disorders in the world. Worldwide, approximately 1.5% of the population, or around 90 million people, are carriers of BT, 400,000 of whom are actually affected.3 According to Orphanet,4 the annual incidence at the birth of symptomatic BT is estimated at 1 in 100,000 worldwide and 1 in 10,000 people in Europe.
It mainly occurs in the Mediterranean countries, but is also found in the Middle East, Central Asia, Southern China, India, South America, and countries alongside the north coast of Africa.5
In the world, the Republic of Maldives has the highest incidence of thalassaemia, with an 18% carrier rate of the population. The estimated frequency of BT in Cyprus is up to 16%; Thailand: 1%; Iran: 5–10%; China: 3–8%; and India: 3–4%.5,6 In Italy, about 7,000 people affected by BT.1 The Piera Cutino Association affirms that there are about 3 million healthy carriers in Italy and 400,000 in Sicily.7
Although BT is relatively rare in the USA, there are an estimated 1.25 million carriers, making up 0.4% of the population. Thalassaemia affects approximately 2,000 patients living in the USA, with 1,000 patients having BT major, but data are largely influenced by factors such as population migration.8
Additionally, it is estimated that the number of newborns affected each year by BT in the countries of the Mediterranean Basin will increase from 2,479 in 2020 to 2,899 in 2050, for annual average growth of 0.5% over 30 years.
THE VALUE OF PREVENTION
Given the prevalence and severity of BT and the cost of treatment, the World Health Organization (WHO) recommends a prevention-based approach.2
The main strategies adopted by national programmes that have shown success and effectiveness include: national policy on prevention that indicates national approval; control and support; public awareness programmes; a screening programme to identify carriers; genetic counselling services; pre-natal diagnosis as a choice for at-risk couples; pre-implantation diagnosis; and new emerging technologies.9
Genetic counselling aims to replace misunderstandings about the causes of genetic disease with correct information, and to increase people’s control of their own and their family’s health by informing them of the resources available for diagnosis, treatment, and prevention. It helps to integrate psychosocial information impacting the family system and relationships, assisting patients in conveying information about genetic risk to other family members, and providing reliable sources of emotional and social support.10
Pregnancy management for females with thalassaemia is more complicated than in females without thalassaemia.11
Although the fetal outcomes for pregnancies achieved by parents with thalassaemia are remarkably successful, there are some important factors (such as cardiac impairment, liver dysfunction, and the vertical transmission of viruses) that must be seriously considered before encouraging a female with thalassaemia to embark on pregnancy.12 Moreover, the highly dynamic state of pregnancy needs higher haemoglobin for better oxygen delivery to the fetus, so females with TDT need transfusions as often as every 2 weeks.11
The aim of these programmes is to allow involved couples to make ‘informed choices’ concerning marriage and reproduction, according to the information provided by the professional offering counselling. The number of married patients and proportion of parents amongst patients with BT could be an index for level of management.11
Prevention has proven effective in Mediterranean countries strongly impacted by the disorder that have opted for this approach, among them Cyprus, Greece, and Italy.13
A combination of implementable structural and functional strategies should help in improving the detection and management of patients with haemoglobinopathies. WHO guidelines recommend that every country should develop, implement, and mandate participation in its own haemoglobinopathy diagnostic and prevention programmes. This would entail planning studies to understand the patterns of mutation in various regions in the country, and then setting up a comprehensive carrier detection programme.2
THE BURDEN OF β-THALASSAEMIA
The serious nature of the disease in its most severe form and its wide diffusion imply a social, psychological, and economic impact on the community and healthcare system.
As in all chronic conditions, patients with BT are more vulnerable to emotional and behavioural problems. BT affects emotional conditions, daily activities, family experiences, and occupational capabilities of patients and their caregivers because of lifelong complicated and burdensome therapeutic protocols.14
Several studies have reported the occurrence of psychopathologies in subjects with TDT, including incidences of social withdrawal, somatic complaints, and social externalising. Attention disorders were present, with major depression and anxiety being the most frequently seen psychological disorders.14,15
In addition, patients with thalassaemia major also suffer from sleep disorders. Sleep disorders could be a result of the short-time and frequent hospitalisation periods these patients have to go through, together with the symptoms caused by anxiety and depression, which can have an infuence on their sleep quality.15
In adult individuals, having thalassaemia represents a burden to their employment and working life. The difficulties experienced range from being discriminated against, having unsupportive colleagues and friends, being inhibited from developing to their full potentials, and getting a lower salary, to being fearful of losing their employment due to their frequent time away from work.16
These significant factors add to the economic burden associated with a chronic disease, with implications for both the individual affected and the healthcare system overall.
In fact, together with medication cost, there are extra costs that include medical consultation, laboratory tests, diagnostic tests, cost of preventative treatments, side effects of therapies, and many other indirect costs. Indirect costs include travel expenses, the cost attributable to the loss of productivity by patients or their caregivers, the impairment of wellbeing, and all other related aspects.17
This aspect becomes particularly pronounced in developing countries, since significant efforts must be invested in improving the medical care of patients with thalassaemia to prolong and improve quality of life (QoL).18
FACTORS INFLUENCING THE SPREAD OF β-THALASSAEMIA
The systematic study of thalassaemias began in 1925, when two paediatricians from the USA, Thomas Cooley and Pearl Lee, presented to the American Pediatric Society (APS) the case of five children who displayed anaemia, an enlarged spleen and liver, low-grade jaundice colouration, and the presence of immature red and white blood cells in the blood.19
Other notable clinical features included skull and facial bone enlargement, skin discoloration, and “conspicuous changes in the long bones,” which led Cooley and his associates to suppose that the symptoms might be due to a congenital malformation of the haematopoietic tissue.19
In addition, by examining several other cases, Cooley realised that patients were frequently of Mediterranean origin (mainly Italians and Greeks); the disease had a strong familiarity (i.e., it was transmitted from parents to children); and bone alterations were often related to particular haematological disorders.19,20
Between 1943 and 1946, Ezio Silvestroni and Ida Bianco demonstrated that thalassaemia major was caused by a hereditary anomaly of the red blood cells that appear smaller than normal, a condition called microcythaemia. With wide population studies, they proved that the disease was extremely frequent in Italy, and that it appeared only in individuals with two parents with thalassaemia, while children were born healthy when only one of the parents showed a thalassaemic trait.19
Once the genetic origin of the disease was recognised, many wondered about its spread, and why it was found mainly in the Mediterranean area. Several factors have contributed to the global distribution of thalassaemias. These factors include malaria resistance, consanguinity, migration, survival rates, and prevention.
Resistance to Malaria
Worldwide, the distribution of malaria and the common haemoglobinopathies have largely confirmed the close relationship between these two pathologies in populations living in highly malarious areas. The relationship that binds these so different diseases, thalassaemia being a genetic condition and malaria an infective disease, derives from the fact that the subject with thalassaemia, whether homozygous or heterozygous, is more unlikely to be infected by malaria than a subject who does not have thalassaemia. According to what Haldane21 affirmed in 1948, thalassaemia, although caused by a genetic defect that in normal environmental conditions is disadvantageous and would be, therefore, suppressed by the genetic selection, has represented instead in an environment afflicted by malaria a notable advantage, and has been able to spread widely. This phenomenon coincidentally facilitated the survival of heterozygous individuals, such as in areas in Italy where malaria was particularly widespread or endemic, including the Po Delta area, Sardinia, Sicily, and Lazio regions.10,12,20,22
Consanguinity
Consanguineous marriage is especially common throughout the Eastern Mediterranean, North Africa, and the Indian subcontinent, where 25–70% of unions involve related family members.23 Religious, cultural, and economic factors are commonly perceived reasons for such marriage. In a 2006 study on consanguineous marriages in Italy, Cavalli-Sforza et al.24 demonstrated that, in some regions of Southern Italy (Basilicata, Calabria, and Sicily), marriages with blood-related partners amounted to over 40% in the years 1935–1939. Some factors such as altitude, village size, population density, and migration have a marked influence on the overall frequency of consanguineous marriages. Increased migratory movements, the changes in communication, transportation, work availability, improved education, and indeed, also the social contacts that occurred in the 20th century, resulted in a rapid decrease in consanguineous marriages.25
Migration
The consistent multi-ethnic migrations of the last decades have considerably changed the epidemiology of haemoglobinopathies. Along with the aforementioned factors, it is also necessary to consider the migratory trends characterising the geographical area of the Mediterranean Basin, which has contributed to the spread of this disease in past centuries up to the present day. In 2017, the global number of refugees reached an all-time high of 25.4 million, including many people from regions where BT is endemic such as Syria, Afghanistan, and Myanmar. In recent years, Italy has accepted many refugees who crossed the Mediterranean Sea, demonstrated by the more than 126,000 applications for asylum submitted in 2017.26
Survival Rates
Thalassaemia used to be a paediatric disease, but the median age of patients has now increased in European Mediterranean countries as a result of increased survival and birth rate reduction. Population screening, genetic counselling, and the availability of pre-natal diagnosis have been extremely effective. In Sardinia, for example, the number of children born with thalassaemia major children per year predicted on the basis of the carrier rate, assuming random mating, shows a reduction from 1:250 live births to 1:1,660 in 2009, with effective prevention in 85% of cases.27 In recent decades, several factors have been shown to improve the survival rate of patients with BT, including implementation of the thalassaemia prevention programmes, increased quality of healthcare services, provision of appropriate treatment, and essential services for these patients.28 According to a recent study, iron chelation therapy resulted in better overall survival of patients with TDT, especially if it is instituted early and compliance is maintained.29 Other factors that improved survival included better awareness of BT and its management among healthcare providers and patients, and guidelines and screening for safe processing of blood and blood products.30
Prevention
Comprehensive prevention programmes include public awareness and education, genetic counselling, and population and carrier screening, accompanied by pre-natal diagnostics, and pre-implantation diagnosis.23,26 The establishment of a national registry allows the census of patients affected by BT, together with data on morbidity, mortality, and other needed therapies, and it constitutes a valuable tool for the evaluation of the efficacy or weakness of the current prevention programmes, and for the planning of prevention and social health interventions.31
CURRENT AND INNOVATIVE THERAPIES
Currently available treatments for the management of patients with TDT and non-TDT include blood transfusion, splenectomy, hydroxyurea, iron chelation therapy, and, for a subset of patients, haematopoietic stem cell transplantation.32
Since blood transfusion is one of the first and most critical components of the clinical management of TDT, it is essential that blood transfusion services in all countries are strengthened, to ensure the availability of a safe and adequate blood supply for all patients who need regular transfusions.2
This issue was severely challenged during the COVID-19 pandemic. The COVID-19 outbreak resulted in the disruption of various aspects of blood supply dynamics. Lockdowns imposed by governments to contain the spread of the virus resulted in reduced movement of individuals, and thus reduced the availability of the blood donors. The fear of acquiring infection during the commute to the blood centre and during the process of blood donation also added to the severely compromised situation. These all led to a sharp fall in the blood collection during initial stages of lockdown, which reduced further as the lockdown progressed.33
The WHO has published interim guidance on maintaining blood supply during the COVID-19 pandemic. It has recommended that blood transfusion services must be prepared to move quickly in response to changes in managing the demand for blood and blood products, while mitigating the potential risk to staff and donors from exposure to COVID-19. In the event of anticipated blood shortages, there should be local strategies to increase supply, prioritise use, review the transfusion threshold of red cell transfusions for patients who are stable and low-risk, and maximise use of alternatives for transfusion.34
Allogeneic bone marrow transplantation can cure TDT, but less than 20% of eligible patients have a related human leukocyte antigen–matched donor.35 An alternative therapeutic approach is represented by gene therapy, which is the current subject of several clinical trials globally.36
Increases in gene editing efficiency, particularly in repair accuracy, will likely translate into an increasing number of clinical applications using gene-edited autologous haematopoietic stem and progenitor cells.
Recently, a therapy currently tested on patients with BT is based upon clustered regularly interspaced short palindromic repeats (CRISPR), one of the most studied examples of genetic editing techniques.
CRISPRs are short DNA sequences with unique spacer sequences that, along with CRISPR-associated (Cas) proteins, constitute an adaptive immune system in many bacteria and archaea against invading bacteriophages. By using short RNA molecules as a template, Cas makes highly sequences specific cuts in DNA molecules that can be exploited to insert genes, or to precisely modify the nucleotide sequence at the cut site.35
The CRISPR-Cas technique presents several advantages: it has been found to be able to modify several genes at once, and it is much more precise in cutting DNA at specific sites, allowing a drastic reduction in off-target cuts.36
In addition, it recognises its target sequence via guide RNA molecules, which can be easily and cheaply synthesised. An ordinary molecular biology laboratory can now edit genes or entire genomes of many organisms, as the CRISPR-Cas system does not require sophisticated knowledge or expensive equipment.37
Therapeutic applications derived from the CRISPR-Cas technique are many and have grown in recent years. Indeed, the annual number of publications on PubMed that have CRISPR in the title and/or abstract increased from 1,000 in 2010 to more than 5,000 in 2020.
To date, only studies on cells and small animals have provided support for the therapeutic efficiency of employing CRISPR-Cas gene-editing technology to rectify pathological mutations that cause genetic diseases. Successful examples include gene rectification in Duchenne muscular dystrophy, sickle cell disease, BT, and hereditary tyrosinaemia Type 1.38
An experimental therapy based on CRISPR-Cas9, CTX001, is currently being studied as a potential one-time therapy for patients with TDT. The study includes 15 patients with TDT who are currently in follow-up to evaluate the safety and efficacy of the therapy, which is already showing reassuring and promising results.35
Luspatercept is the most recently approved agent (in the USA and Europe) for the treatment of adults with TDT.39 It is a recombinant fusion protein that binds to select transforming growth factor β superfamily ligands and enhances late-stage erythropoiesis.40 A Phase III, randomised, placebo-controlled trial established the efficacy and safety of Luspatercept in reducing the transfusion burden among patients with TDT. The results of all primary and key secondary efficacy analyses were in favour of Luspatercept over placebo. Furthermore, a greater percentage of patients in the luspatercept group than in the placebo group had reductions in the transfusion burden of at least 33%, or at least 50% from baseline during any 12-week or 24-week interval. Now, a 5-year open-label extension Phase is underway to provide long-term data on the safety of Luspatercept, and its effects on the transfusion burden and iron outcomes.40
METHODS
The difficulties related to this pathology, which presents a serious clinical framework in its major form, and the absence of definitive therapies make it a burden at an economic as well as a social level. The latter, in fact, is more difficult to address and assess compared to the economic impact due to the disease. Trying to give an answer to how the disease has an impact on the lives of people affected, the authors have conducted a review of the literature on the subject.
Electronic databases including PubMed, Scopus, and Web of Science were used to search for the articles. Related article titles were selected and reduced to the abstracts of the relevant articles, after which the 14 selected full articles were reviewed. Searches for literature on the topic were carried out using these electronic databases and also integrated with a free search on web-based search engines.
These databases were chosen based on the usage of previous similar studies. To capture the wide array of studies that may be relevant to this topic, the authors did not pre-define the study designs of included studies.
All types of study design, such as qualitative and quantitative studies, were included in the search. The selected years range of the publications’ search was limited to studies from the year 2010 to the date of writing, because of the fast-paced field of innovations of genetic therapies for the treatment of BT driving attention to this disease, and the resulting most current publications on the topic. In addition, the research focused on the contribution of articles in both Italian and English languages (both being known by the authors). Geographical location limitations were not applied.
Keywords including: ‘beta thalassemia’, ‘beta thalassemia major’, and ‘transfusion-dependent thalassemia’, combined with the terms ‘social impact’ or ‘social burden’, were used to achieve relevant studies. A search was completed using AND and OR to combine the results that were found based on each keyword.
The exclusion criteria regarding other works concerned, publications written in languages other than English and Italian, PhD theses, grey literature, and reports. The search resulted in 14 relevant articles that allowed for a discussion of the meaning of social impact of BT, and the possible solution aimed at better advocacy and awareness of this disease.
A total of 79 article titles related to the topic came up in the search. From these titles, 57 studies, which were more closely related to the purpose of the review, were shortlisted and their abstracts were screened. From this, 21 studies were eligible for full-text screening. After thoroughly reading the articles and excluding those that did not directly address the significance and an assessment of the social impact of BT, the final number of articles included in the review was 14. The Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) flow diagram below (Figure 1) lays out these procedures in more detail.41
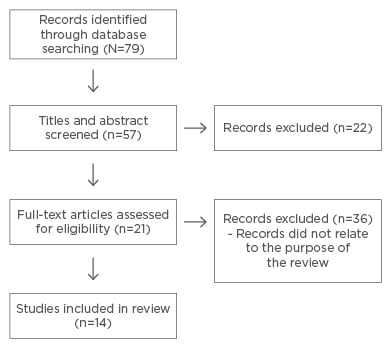
Figure 1: Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) flow diagram.
Table 1 shows the selected papers, indicating the key words, main findings, and the authors’ geographical location.
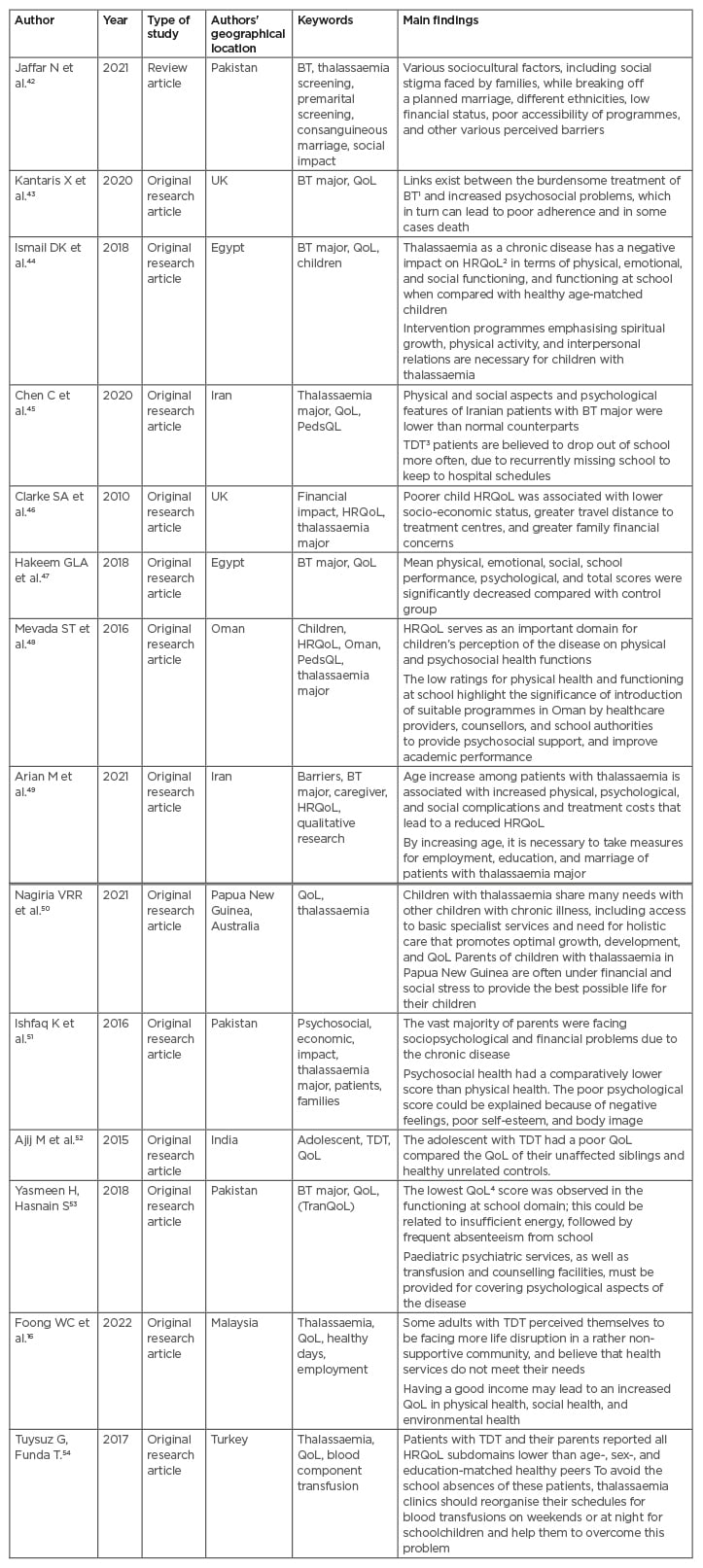
Table 1: Reviewed papers.
BT: β-thalassaemia; HRQoL: health-related quality of life; QoL: quality of life; PedsQL: paediatric quality of life inventory; TDT: transfusion-dependent β-thalassaemia; TranQoL: Transfusion-dependent quality of life questionnaire.
RESULTS
The results of the review show similar outcomes about the psychological and social burden patients face. It is interesting to remark upon the geographical area of origin of the authors of the studies proposed here, since most are areas where there is a relatively higher prevalence of patients with BT. This consequently allows authors to design studies on the subject, compared with those in other realities that are geographically further away, or that do not experience a similar prevalence of the disease. The limited geographical distribution of BT, along with its rarity, affects the number of studies published on the social impact of the condition, as it limits the number of patients that can be enrolled in studies.
The results of this review show that 78.5% of the selected studies use health-related QoL (HRQoL) as a method of assessing the social impact of BT. HRQoL is a multidimensional concept that includes three broad domains (physical, psychological, and social functioning), which may be affected by an illness and/or its treatment. QoL measurements are increasingly considered important when evaluating disease progression and treatment.55
The use of questionnaires and interviews for the assessment of the social impact of BT were found in the studies reported in the review. The WHO Quality of Life questionnaire in its abbreviated version (WHOQOL-BREF) was found to be used in two studies; one study used the Transfusion-dependent QoL questionnaire (TranQoL); five studies used the Paediatric Quality of Life Inventory (PedsQL) 4.0 measurement model; one study used the Thalassaemia Life Index (ThALI); three studies used questionnaires and interviews approved by the ethics committee of the referring institution where the study was conducted.
The self-reported WHOQOL-BREF questionnaire is a short version of the WHO Quality of Life 100 (WHOQOL-100) questionnaire. The WHOQOL-BREF was developed to provide a quick assessment tool for QoL, and it assesses the individuals’ perceptions of their position in life in the context of the culture and value system in which they live, and in relation to their goals, expectations, standards, and concerns.52
TranQoL is the first disease-specific instrument that measures HRQoL in patients with TDT. It has four versions: a child self-report; an adult self-report; a parent self-report (measuring the impact of the disease on the parent); and a parent proxy-report (measuring the child’s QoL). The adult and parent self-report questionnaires include a fifth category on sexual activity, which represents only one item.56
PedsQL 4.0 measures the essential core domains for paediatric HRQoL. It encompasses the essential core domains for paediatric HRQoL measurement, which are physical functioning, emotional functioning, social functioning, and functioning at school. It consists of appropriate forms for ages 2–4, 5–7, 8–12, and 13–18 years.44,54
ThALI addresses the multidimensionality of QoL, and encompasses different items such as general physical health, coping, body image, appearance and confidence, social relationships, and autonomy.43
If semi-structured interviews and questionnaires are chosen as a method of assessing the social impact of BT, they must be validated by the ethics committee responsible for the centre in which the study is being conducted, in order to protect the rights, dignity, and integrity of the patients involved, and to adapt the study protocol to meet national standards and guidelines.49
HRQoL in children with chronic diseases can be adversely affected as a result of hospital appointments, restricted activities, and general worries.44,55
The results indicate consistent agreement in observing that the health and social scores obtained from the various investigations carried out on patients with BT are considerably lower compared with the healthy population. The challenges faced by these patients affect all aspects of their lives economically, physically, socially, and educationally.
The lowest scores, associated with a poorer QoL in patients with BT, are found in the domain of physical and psychological functioning,45,49,52,54 together with a lower score observed in the functioning at school domain that could be related to insufficient energy followed by frequent absenteeism from school, which had a negative impact on QoL.44,47,48,53
Outcomes may also vary depending on a patient’s family’s economic background and geographical location. In middle- to low-income countries, in addition to economic aspects, religious factors and social stigma also play a role in lowering the level of QoL.16
DISCUSSION AND CONCLUSIONS
BT is a rare genetic disease that currently lacks a definitive cure. In its most severe form, BT major, the disease involves frequent lifelong blood transfusions and other complementary therapies that greatly affect patients’ lives economically, psychologically, and socially.
The rare nature of the disease hinders the research for a definitive cure, despite recent technological advances and promising results from ongoing trials.35,40
A crucial contribution is provided by prevention programmes such as population screening, parental education, pre-natal diagnosis, and genetic counselling,51 which support raising awareness of the severity and complications of the disease and have been successfully implemented in several disease- prone areas worldwide.9
This review herein highlights how the social dimension of patients with TDT is very neglected in studies on the disease, both because as a rare disease its prevalence is low and, therefore, it is difficult to include patients in this type of study, and because the concept of social impact is difficult to analyse and evaluate.
Most of the studies have calculated the social impact of BT using the HRQoL. The QoL of people with BT resulted, in all domains considered, lower than healthy people representing the control group. As a chronic disease, BT has a negative impact on HRQoL in terms of physical, emotional, and social functioning, and functioning at school. This highlights the significance of the introduction of suitable programmes by healthcare providers, counsellors, and school authorities to provide psychosocial support and improve academic performance.48
It is essential to address these outcomes and try to fill the gaps in the psychological support and social introduction of these patients, a goal that can be achieved with a robust network of prevention programmes.
Psychosocial support and family therapy are considered as essential aspects of care management to promote HRQOL in patients with chronic disease. Promoting community knowledge and planning for the presence of these patients in the community should be included in the healthcare policies for these patients.49
Educatory intervention could help to improve educational levels in patients with BT, and positively affect the social functioning domain.45 This concern needs to be reviewed by health providers, families, and educational services.
Moreover, as the timing of screening is an important determinant in the success of screening programmes, it is recommended that the test should be performed at an early stage (prior to engagement), when it is easier for people to make a decision about their marriage, and their desire for parenthood. For this purpose, health education needs to be aimed at adolescents to change their opinion regarding genetic counselling and ensure that this educational process becomes properly structured and effective in society.42
Parallel with age increase, it is necessary to plan for patients’ presence in society, and specific measures should be taken for employment, education, and marriage of patients with thalassaemia major.49,57,58 Intervention programmes emphasising spiritual growth, physical activity, and interpersonal relations would have a significant impact on patients with thalassaemia.





