Interview Summary
Pyruvate kinase (PK) deficiency is a hereditary haemolytic anaemia caused by mutations in the PKLR gene encoding PK, which is critical for maintaining red blood cell (RBC) energy levels. Defects in PK cause chronic haemolysis. There are currently no disease-modifying therapies approved for use in children with PK deficiency, and treatment can involve regular blood transfusions, iron chelation, splenectomy, and management of disease complications. In this interview, Rachael F. Grace, a paediatric haematologist at the Dana–Farber/Boston Children’s Cancer and Blood Disorders Center, Massachusetts, USA; and Julián Sevilla, a haematologist at the Hospital Infantil Universitario Niño Jesús in Madrid, Spain, shared their experience of diagnosing and treating paediatric patients with PK deficiency. They discussed the substantial variability in symptoms between patients irrespective of their haemoglobin level, the risk of iron overload even in children not receiving regular transfusions, and the effects of jaundice on children’s self-esteem.INTRODUCTION
PK deficiency is an under-recognised, hereditary, non-spherocytic haemolytic anaemia, caused by mutations in the PKLR gene encoding PK, which is critical for maintaining RBC energy levels and morphology. Defects in PK cause chronic haemolysis, which leads to other long-term complications.1,2
PK deficiency affects an estimated three to eight per million people,3 though the true frequency is unknown because of the propensity for rare conditions to be undiagnosed or misdiagnosed. The clinical symptoms and findings of PK deficiency can vary substantially between individual patients, from a life-threatening disorder at birth, to mild symptoms that may lead to a diagnosis later in childhood or adulthood.1
Management is supportive, and includes blood transfusions in patients who experience significant symptoms of haemolytic anaemia, and consideration of splenectomy. There are currently no disease-modifying therapies approved for use in children with PK deficiency, though research is being conducted into PK activators and gene therapy.1,4-7
SYMPTOMATIC VARIATION BETWEEN PATIENTS WITH PYRUVATE KINASE DEFICIENCY
Sevilla explained that it is impossible to make a generalisation regarding how the life of a child with PK deficiency might differ from that of a healthy child, because of the wide variation in disease severity. He emphasised that there is a vast difference between a patient who comes into the hospital every few weeks for a transfusion, and one who is not receiving transfusions, and comes into the clinic for an occasional check-up.
Grace agreed that the spectrum of clinical manifestations she sees in her clinic is broad. She stressed that this may make it difficult for clinicians who are less familiar with PK deficiency to recognise and diagnose the condition in their patients.
Symptoms and impact on routine and daily functioning are highly variable. Children may be on chelation medication to treat iron overload, which can occur both in children who are regularly transfused and those who are not. Children with PK deficiency require regular monitoring, including regular blood draws to monitor their haemoglobin and iron levels. Some patients with PK deficiency may be active, and may not notice their anaemia day-to-day. Grace explained that symptoms such as fatigue or lack of energy are chronic in patients with PK deficiency, and these children will have effectively experienced them since birth, so they may be unaware of them during everyday life. Children with intact spleens may have splenomegaly,1 which might require modifications to daily activities, such as avoiding contact sports, to avoid the risk of splenic injury.
Grace also described a subgroup of children that experience significant disease-related fatigue, which affects the energy and concentration they have at school and at after-school activities.
RELATIONSHIP BETWEEN HAEMOGLOBIN LEVELS AND SYMPTOMS
In general, the relationship between haemoglobin and symptoms among patients with PK deficiency is not consistent; however, there is often a relationship between haemoglobin level and symptoms such as fatigue in individual patients.8 Although the reason why patients experience different symptoms at the same haemoglobin level is not fully understood, haematologists manage patients with PK deficiency based on an individual patient’s symptoms, rather than haemoglobin level alone.
Sevilla described a highly functioning adult patient who exercises regularly, who was surprised to discover how much better he felt following treatment of his underlying disease with an oral PK activator. Conversely, Grace explained that her clinic sees children with PK deficiency who receive transfusions to raise their haemoglobin levels, but who may still experience symptoms between transfusions, such as low energy and difficulty concentrating in school.
SYMPTOMS AND COMPLICATIONS BEYOND ANAEMIA
The symptoms of PK deficiency include jaundice (yellow skin/eyes), fatigue, and shortness of breath. Signs and symptoms experienced by patients with this condition are summarised in Figure 1.
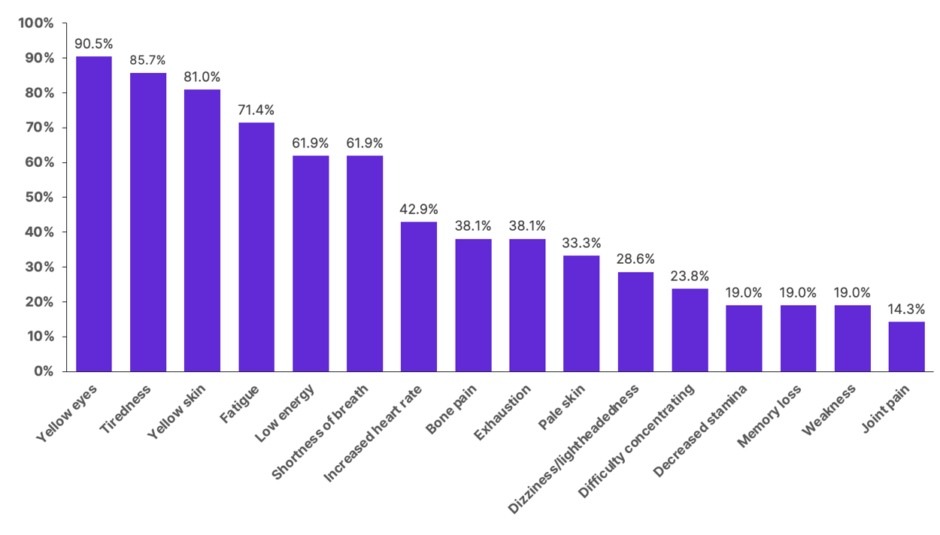
Figure 1: Signs and symptoms reported by patients with pyruvate kinase deficiency.
Adapted from Grace et al.9
Chart includes signs and symptoms reported by ≥3 adults with pyruvate kinase deficiency (n=21).
Patients with PK deficiency can develop complications, regardless of whether they are regularly or not regularly transfused. Exposure to transfusions increases the risk of transfusion-related complications, such as transfusion reactions, iron overload, alloimmunisation, and infection.1,10 For patients with an indwelling catheter, they incur risk of catheter-associated infection and thrombosis.
Iron Overload
It is well known that patients receiving regular blood transfusions can develop iron overload, a condition associated with significant morbidity.11 However, Sevilla explained that iron overload can also occur in patients with PK deficiency who do not receive regular transfusions, due to increased intestinal iron absorption.11,12 Since iron overload is associated with multiple complications, including liver cirrhosis, cardiac disease, and endocrinopathy, and it is not associated with symptoms until a significant amount of iron is deposited, regular monitoring is important to allow for timely and adequate chelation treatment.1
Gallstones
Bilirubin accumulation leads to the development of gallstones in nearly half of patients with PK deficiency.10,11 Similar to iron overload, Sevilla stressed that gallstones are more common in children that have significant haemolysis, but they also occur in children with less haemolysis. He suggested that the risks of both iron overload and gallstones are often under-recognised in children who are not regularly transfused, as they tend to visit the clinic less often than those who are regularly transfused.
Viral Infections
Grace explained that any child who develops a viral infection can experience increased haemolysis,13 and in this setting, a patient with PK deficiency may have a longer recovery time than a patient without PK deficiency. They may also require a blood transfusion in this setting, particularly if an aplastic crisis arises.13
Jaundice
The degree of jaundice or scleral icterus in PK deficiency depends on a combination of the level of haemolysis in each patient, and their genetic ability to metabolise bilirubin. Some patients with PK deficiency also have Gilbert’s syndrome, a common inherited condition which affects the processing of bilirubin in the liver.13 Sevilla stressed that high bilirubin levels may have a greater impact on these patients’ lives than their haemoglobin level. They may have normal haemoglobin levels, but an increased risk of gallstones,10 and extremely yellow skin. Grace agreed that for these patients, their yellow appearance is a major problem (Figure 1), with many experiencing bullying or unwanted curiosity from strangers. This can have a substantial effect on the patient’s self-esteem and quality of life, particularly in adolescence, and the impact of jaundice should not be underestimated.
Because of the risk of kernicterus associated with high levels of bilirubin in infants, any newborn with jaundice receives aggressive treatment1 to reduce bilirubin levels, including phototherapy and an RBC transfusion if necessary. Sevilla explained that such treatment is performed in the neonatal intensive care unit without the need to understand the root cause of the condition, and haematologists will often only be consulted if the jaundice persists.
DIAGNOSTIC CHALLENGES
One of the major diagnostic challenges that Sevilla and Grace described was the very early step of considering enzymopathies, such as PK deficiency, when a child presents with haemolytic anaemia. They indicated that clinicians generally consider RBC membrane defects as a potential cause first, as these are more prevalent conditions than PK deficiency. In addition, Grace suggested that it is not always clear that PK deficiency could be the diagnosis, because the presentation can be so variable.
If a clinician considers haemolytic anaemia in a child to be due to hereditary spherocytosis, perhaps because family members were diagnosed with this in the past, they may not conduct further tests, and this can lead to a misdiagnosis. However, Sevilla stressed that these family members will have been diagnosed many years ago, when there were no tools to identify a disease like PK deficiency, and this should not be the case today.
Sevilla explained that the eosin 5-maleimide binding assay, a simple test used to diagnose the most frequent hereditary haemolytic anaemia, hereditary spherocytosis, can help physicians to rule out this disease, and to start considering alternative causes. Testing for PK levels in RBCs can be difficult, and it requires a high level of expertise. Sevilla emphasised that, in Spain, this test is only performed in specialist laboratories. Diagnosis through genetic testing for mutations in PKLR is often easier, since genetic testing facilities are more abundant.
Grace expanded on this by suggesting that much has been written about the partnership between genetic and enzyme tests for PK deficiency, and ideally performing both tests together can provide the most information.10,13 She explained that because there are so many genetic variants of PKLR, it is not always clear whether a patient’s specific variant is disease-causing. Though certain genotypes (those homozygous for non-missense mutations, or for missense mutations that affect the PK enzyme’s active site, or overall protein stability) are associated with a more severe phenotype, there is significant genotype-phenotype discordance in PK deficiency, such that any individual patient’s phenotype cannot be predicted with certainty from genotype alone.14
Both Sevilla and Grace stressed that, for patients with mild haemolytic anaemia, clinicians may feel there is no need to continue testing to identify the root cause. These are patients that may go undiagnosed. With an increased understanding of the need for monitoring in PK deficiency, and the potential for new treatments, it is important to diagnose all patients with PK deficiency, even those with mild haemolysis. Grace recommended that clinicians communicate with medical centres that have expertise in RBC disease, because their laboratories can be extremely helpful in terms of making a diagnosis, and they can provide expert opinion about management following a diagnosis. This is also paramount for genetic counselling.
MANAGEMENT CHALLENGES
Clinically available and research-based management strategies for children with PK deficiency are summarised in Figure 2. Grace stressed that there are enormous challenges for haematologists in treating children with PK deficiency. She described that one of the most critical issues for paediatric patients with PK deficiency is the lack of approved disease-modifying therapies, other than allogenic haematopoietic stem cell transplantation. The available treatments are supportive, addressing the symptoms that impact a patient’s quality of life. However, both transfusion and splenectomy are associated with potential complications: transfusion causes iron overload, and patients need chelation therapy, while permanent venous access and splenectomy are both associated with an increased risk of infection and thrombosis.12 Grace explained that it is unclear which patients will respond well to splenectomy, and whether this is the optimal long-term management approach.
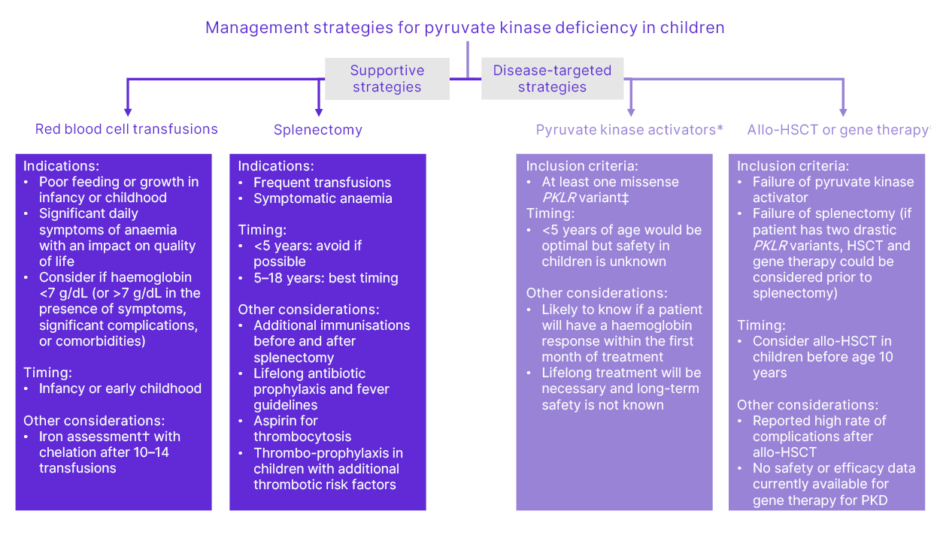
Figure 2: Clinically available and research-based management strategies for children with pyruvate kinase deficiency.
Adapted from Grace et al.13
*Treatments currently being evaluated in clinical trials. See the EU Clinical Trials Register for more information (https://www.clinicaltrialsregister.eu/). The safety and efficacy of PK activators and gene therapy in paediatric PKD are under investigation, and have not been established. There is no guarantee that mitapivat or gene therapy will receive health authority approvals or become commercially available in any country for the uses under investigation.
†Iron overload often occurs in the absence of transfusion. Chelation therapy should be strongly
considered if liver iron concentration >5 mg/g or ferritin >800 µg/L and transferrin saturation >60%,
even in the absence of transfusions.
‡Future studies may indicate that other genotypes are responsive.
Allo-HSCT: allogenic haematopoietic stem cell transplantation; PK: pyruvate kinase;
PKD: pyruvate kinase deficiency.
Even in those patients who receive regular transfusions, it can be difficult to determine the optimal frequency and volume for the transfusions, and the lowest haemoglobin that is acceptable in patients. For example, Grace described the difficulty of determining the transfusion needs of an infant with haemolytic anaemia. Since all babies experience a natural decrease in haemoglobin levels during the first few months of life, it can be hard to discern whether a transfusion is needed. She stressed that if you transfuse an infant with PK deficiency in early life, it becomes difficult to assess the transfusion needs as they age. The challenge lies in evaluating the level of intervention needed to maintain good growth and development, without overtreating.
Once a child reaches 5 years of age, splenectomy becomes an option for PK deficiency treatment.12 Sevilla explained that for some patients, the family is already aware that a splenectomy is likely to be needed, and may be awaiting this procedure at age 5. However, there are also patients for whom is it less clear whether splenectomy is the right approach, and parents may also be uncertain because it is not effective in a subset of patients, and because of the potential for long-term complications.12
Grace stressed that there are many aspects of paediatric PK deficiency management that are not fully understood. Because PK deficiency is a rare disorder, carefully described cohorts and registries are used to better understand how best to use the available management strategies.
In addition, Grace explained that when considering whether to enrol patients in a clinical trial for an investigational treatment, it is important to weigh up the effects of school absence, and the benefit/risk profile of each therapy with the current impact of the disease on a patient’s quality of life.
UNMET NEEDS IN CHILDREN WITH PYRUVATE KINASE DEFICIENCY
While it may be clear that patients who are regularly transfused require monitoring and chelation therapy, Grace expressed concern that patients who are not regularly transfused may not always receive the attention they need from their medical team. Sevilla noted that although his regularly transfused patients would, understandably, prefer not to have to go the hospital for frequent transfusions, these patients are seen more frequently compared to the not regularly transfused population. The not regularly transfused population attends the clinic less regularly, which can sometimes mean these patients receive less specialised care. Monitoring recommendations for children with PK deficiency are summarised in Figure 3.
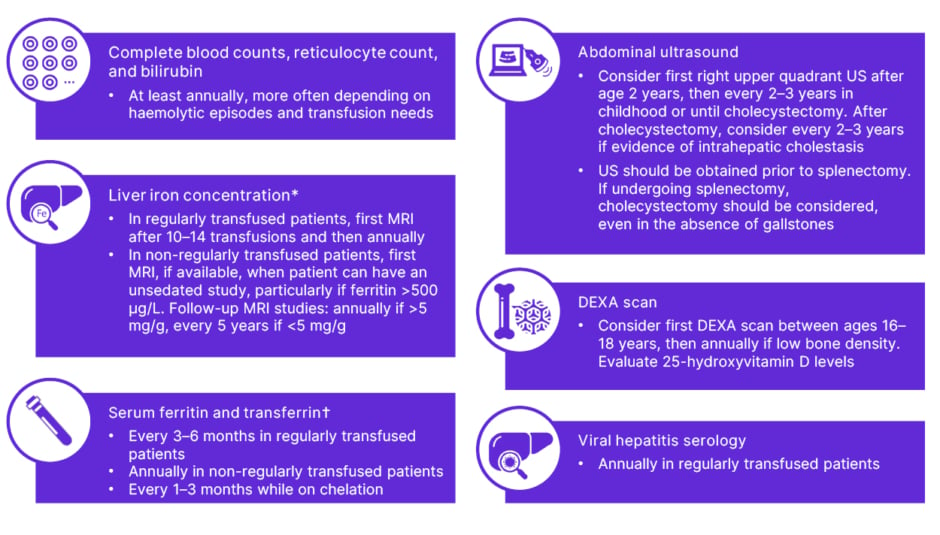
Figure 3: Monitoring recommendations for children with pyruvate kinase deficiency.
Adapted from Grace et al.13
Regularly transfused patients (≥6 transfusions per year); non-regularly transfused patients (<6 transfusions per year).
*Determined by T2-MRI.
†Iron overload can be defined as liver iron concentration >5 mg/g or serum ferritin >800 mg/L and transferrin >60% (if T2-MRI not available).
DEXA: dual-energy X-ray absorptiometry; US: ultrasound.
Grace and Sevilla emphasised that patients who are not regularly transfused often experience manifestations of their disease that impact their quality of life, and, just like regularly transfused patients, they are at risk of complications.
Therefore, these patients need to receive regular monitoring, and to visit the clinic regularly to discuss whether treatments should be considered.
PK deficiency is a rare disease, and Grace emphasised that this means it can be challenging to conduct sufficient research into the complications that can occur, and that more needs to be done in this area. Evidence-based management guidelines are in development, which will help to clarify which treatments and monitoring plans are best for each patient. Sevilla stressed that research conducted
with PK deficiency cohorts in the past 10 years15 have led to a much clearer understanding of the natural history of the disease, which can inform better management overall.
New treatments for PK deficiency are needed, which could improve energy levels and quality of life in these patients, perhaps even permitting a cessation of transfusions, and/or avoidance of splenectomy.
THE FUTURE OF PAEDIATRIC PYRUVATE KINASE DEFICIENCY TREATMENT
Sevilla explained that considerable research has been conducted into PK deficiency over the past 20 years, and there are several clinical trials currently assessing new treatment options for these patients.
Allogenic haematopoietic stem cell transplantation is one potential curative therapy currently available for PK deficiency. Only a small number of patients with PK deficiency who have undergone stem cell transplant have been reported. In these reported patients, there was substantial toxicity, particularly in children over the age of 10 years.16 However, Sevilla stressed that this treatment should still be considered as a potential treatment option early in life.16
Following two successful Phase III studies in adults, paediatric clinical trials were initiated to investigate the use of the PK activator, mitapivat, in children with PK deficiency.3-6 Mitapivat increases the activity of PK, including a range of mutant forms of PK found in RBCs of individuals with PK deficiency, thereby increasing energy production. It is the first therapy for PK deficiency that potentially targets the underlying metabolic defect that causes the disease. These Phase III trials are investigating the effect of mitapivat on haemoglobin levels for not regularly transfused patients, and transfusion burden for regularly transfused patients, in paediatric patients with PK deficiency (NCT05175105 and NCT05144256).6,7
The use of gene therapy for the treatment of PK deficiency in adults and paediatrics is also currently under investigation in a Phase I clinical trial.17,18 Initial data from this trial, using a self-inactivating lentiviral vector, indicate that this approach is well-tolerated, with no serious adverse events reported in adults (>3 years of follow-up) or children (<1 year of follow-up). The data suggest that this therapy may normalise haemoglobin, improve health-related quality of life metrics, and/or reduce transfusion. Adult patients showed polyclonal reconstitution of their haematopoiesis, with no evidence of clonal evolution, and similar results were reported in paediatric patients, although one of these patients experienced a more limited haemoglobin increase.17,18 Sevilla explained that these promising results are paving the way for a Phase II trial, which is expected to begin very soon.
ADVICE FOR PAEDIATRIC PATIENTS AND THEIR CAREGIVERS
Grace emphasised the importance of two-way communication between a haematologist and a patient and their family, with the haematologist sharing information regarding monitoring, potential manifestations, and management from a medical perspective, and a family sharing how PK deficiency impacts their child, and their specific needs. She also stressed that, although it can be difficult, families should feel confident and comfortable advocating for their child.
Since PK deficiency is a rare condition, families may know more about the condition than their haematologist, at least initially, and she encouraged them to bring in any medical papers they have come across for discussion, or to ask whether their child is eligible for a clinical trial.
There are also patient groups for patients with PK deficiency and their families, with websites or social media presence, which can be excellent sources of information, such as Thrive with PK deficiency,19 and the Pyruvate Kinase Deficiency Foundation.20 Sevilla particularly encouraged the families of children who are not regularly transfused to connect with these groups, as they often have less contact with their haematologist. Patient groups may provide a means for patients and their caregivers to understand potential complications of the disease, and to recognise that they may need to consider treatment to reduce their risk of developing these complications.
CLOSING REMARKS
Grace and Sevilla are hopeful about the future treatment landscape of PK deficiency in paediatric patients. Over the next 5–10 years, ongoing clinical trials investigating disease-modifying therapies have the potential to expand upon the treatment options that are available today.





