
Abstract
Pernicious anaemia (PA) is an autoimmune disease of multifactorial aetiology involving environmental and immunological factors. It is the most common cause of cobalamin deficiency anaemia worldwide. The disease is a macrocytic anaemia caused by a vitamin B12 deficiency, which, in turn, is the result of intrinsic factor deficiency, a protein that binds avidly to dietary vitamin B12 and promotes its transport to the terminal ileum for absorption. Despite the advances in understanding the pathogenesis and molecular biology, diagnosis of PA is still challenging for clinicians because of its complexity, diverse clinical presentations, and the limitations of the available diagnostic tools for the evaluation of cobalamin status and the presence of chronic autoimmune atrophic gastritis. Asymptomatic autoimmune gastritis, a chronic inflammatory disease of the gastric mucosa, precedes the onset of corpus atrophy by 10–20 years. Diagnostic dilemmas could occur when patients with PA present with spuriously normal or high cobalamin levels, normocytic or microcytic anaemia, nonanaemic macrocytosis, autoimmune haemolytic anaemia, pseudo-thrombotic microangiopathy, hyperhomocysteinemia-associated thromboembolism, pseudoleukemia, bone marrow failure, and neurologic manifestations without anaemia or macrocytosis. Other autoimmune disorders, especially thyroid disease, Type 1 diabetes mellitus, and vitiligo, are also commonly associated with PA. The present review focusses on novel aspects regarding the pathogenesis, clinical presentation, and the diagnostic approach of PA; the true usefulness of serum vitamin B12 levels; and the risk of adenocarcinoma and gastric carcinoids as well as their treatment and monitoring strategies.
INTRODUCTION
Pernicious anaemia (PA) (also known as Biermer’s disease1 and Addisonian anaemia2) is a macrocytic anaemia caused by a vitamin B12 (cobalamin) deficiency, which, in turn, is the result of intrinsic factor (IF) deficiency. IF is a glycoprotein that binds cobalamin and thereby enables its absorption at the terminal ileum.3 Whether the stomach pathogen Helicobacter pylori plays a causative role in PA is unclear. The deficiency of IF is a consequence of the presence of atrophic gastritis, which results in the destruction of the oxyntic mucosa and thus the loss of parietal cells, which normally produce hydrochloric acid as well as IF.4 Acid loss leads to iron deficiency anaemia that precedes cobalamin-deficient PA by many years. PA is widespread across all continents and the prevalence of the disease ranges from 50 to 4,000 cases per 100,000 persons, depending on the diagnostic criteria.5 However, PA prevalence is probably underestimated as a result of the complexity of the diagnosis. The incidence of PA increases with age and is rare in people <30 years of age. The mean age of patients with PA ranges from 59 to 62 years. PA is more common in people with African or European ancestry than in those with Asian ancestry. The highest prevalence is seen in northern Europeans, especially those in the UK and Scandinavian countries.6,7 The symptomatology is dominated by a profound megaloblastic type anaemia and, in the most serious cases, by neurological alterations, which can precede the diagnosis of gastric atrophy by several decades. Furthermore, PA correlates with other autoimmune diseases (thyroiditis and Type 1 diabetes mellitus) as well as a genetic disease (genotypes HLA-DRB1*03 and DRB1*04);8,9 however, differential diagnosis may sometimes be challenging because of the limitations of currently available diagnostic tools.
PATHOPHYSIOLOGY
Physiological Roles of Vitamin B12
Vitamin B12, or cobalamin, is an hydrosoluble vitamin synthesised by micro-organisms and detected in trace amounts mostly in foods of animal origin. The normal daily cobalamin requirement for an adult corresponds to 1–2 nmol/L. The absorption and transport mecchanisms are dependent on three key proteins: haptocorrin (HC), IF, and transcobalamin (TC).
In the stomach, vitamin B12 released from food protein by peptic action is bound to HC and travels to the duodenum, where pancreatic proteases digest HC, releasing vitamin B12 to bind to IF. The IF–vitamin B12 complex binds to a specific receptor called cubilin in the distal ileum and is internalised. Eventually it is released by lysosomes, and transported into the blood. Both HC and TC bind circulating vitamin B12 and although the latter is the cellular delivery protein, it is transported together with TC I, II, or III to finally be stored in the hepatocyte.4,5
Cobalamin acts as a fundamental enzymatic cofactor in myelopoiesis (role in nucleotide synthesis) and myelination of the central and peripheral nervous system. The two enzymes through which vitamin B12 serve this function are methionine synthase and methylmalonyl-CoA mutase. Thus, cobalamin serves as a cofactor for methionine synthesis through the transfer of a methyl group to homocysteine which is an atherogenic and potential endothelial toxin. This conversion of homocysteine to methionine forms demethylated tetrahydrofolate which is required for DNA synthesis. Further metabolism of methionine to S-Adenosyl methionine is essential for myelin synthesis and the maintenance of neuronal integrity, as well as for neurotransmitter regulation. Thus, a lack of cobalamin leads to either the destruction of myelin sheaths or the incorporation of abnormal fatty acids into myelin sheaths, thereby leading to impaired neural function and/or transmissions which may be the underlying cause of the neurological symptoms seen in vitamin B12 deficiency.
The resulting anaemia may be macrocytic with bone marrow promegaloblastosis, reflecting ineffective erythropoiesis, or normocytic reflecting concomitant iron deficiency from achlorhydria.
Dyssynchrony between the maturation of the cytoplasm and that of the nuclei leads to macrocytosis, immature nuclei, and hypersegmentation in granulocytes in the peripheral blood. The ineffective erythropoiesis results in intramedullary haemolysis and the release of lactate dehydrogenase (LDH).4,8 Inhibition of DNA synthesis as a result of vitamin B12 deficiency causes megaloblastic changes not only in bone marrow but also in other rapidly dividing cells, such as gastrointestinal epithelial tissue, explaining gastrointestinal disorders in patients with PA.
Physiopathology of Pernicious Anaemia
PA is a complex, autoimmune, multifactorial disease. The environment appears to play a crucial, independent role in the pathogenesis of PA. Even though PA associated with gastric atrophy is now considered an outcome of chronic H. pylori infection, the relationships between PA and H. pylori is still not clear with conflicting views.10-14 H. pylori are ubiquitous organisms invading the gastric mucosa and are a global burden. They cause superficial gastritis, destruction of gastric parietal cells, and atrophic gastritis, resulting in reduced availability of IF for vitamin B12 transport. This causes an interference with vitamin B12 absorption, thus leading to vitamin B12 deficiency and its clinical manifestations. In genetically susceptible individuals, H. pylori infection triggers autoantibodies by a mechanism of molecular mimicry;12,13 however, studies showed that only a minority of patients with PA are infected with H. pylori.12,13 The effect of H. pylori treatment on the potential reversal of atrophic gastritis is also controversial.14,15
Studies to understand the genetic component of PA are long overdue and may provide important insights into its mechanism. Furthermore, rapid progress has been made in the understanding of susceptibility to a spectrum of other autoimmune diseases through genome wide association studies.
There are two key facts that confirm the existence of a genetic basis. Firsty, PA has a familial link with ≤19% of the patients having a family member with PA. On the other hand, it has been observed that the genotypes HLA-DRB1*03 and DRB1*04 were significantly associated with PA. These genotypes were seen in other autoimmune diseases and support the concept that autoimmunity may play a role in PA.16-18
Patients with PA have been shown to have two types of antibodies: one to parietal cells and the other to IF (IFA) or its binding site in the small bowel. The immune response is directed against the gastric H+/K+-ATPase, which accounts for the associated achlorhydria. This proton pump is responsible for acid secretion in the stomach and is the major protein of the secretory canaliculi of gastric parietal cells. It produces acid by secreting H+ ions in exchange with K+. The gastric H+/K+-ATPase is formed by a catalytic 100 kDa α subunit and a 60–90 kDa β subunit. The highly conserved catalytic α subunit is phosphorylated during its reaction cycles and the β subunit comprises a heavily glycosylated 35 kDa core protein. The atrophic gastritis is caused by the action of autoreactive CD4+ T cells that recognise H+/K+-ATP ase, which leads to their immune destruction.19-21 The β subunit is considered the causal antigen and the source of the autoimmune response responsible for the damage to the gastric mucosa. Parietal cells are present at a high frequency in PA (80–90%), especially in early stages of the disease and bind to both the α and β subunits of gastric H+/K+-ATPase. In the later stages of the disease, the incidence of parietal cells decreases due to the progression of autoimmune gastritis and a loss of gastric parietal cell mass, the result of the decrease in antigenic rate.
Studies have reported IFA positivity in 40–60% of patients with PA. These antibodies lead to cobalamin malabsorption in the terminal ileum that leads to cobalamin deficient megaloblastic PA.22,23 However, iron deficiency anaemia, a known complication of achlorhydria, occurs predominantly in women and precedes the onset of cobalamin-deficient PA by approximately 20 years.19,24 So, the patients with unexplained iron deficiency anaemia should be checked for autoimmune gastritis and PA.
CLINICAL FEATURES
PA usually manifests itself in people >30 years old (usually adults >60 years) and affects both sexes equally. The clinical presentation proceeds gradually, and patients usually exhibit symptoms of anaemia with pallor, fatigue, lightheadedness, or tachycardia and decreased mental concentration. Involvement of small-bowel epithelium may result in malabsorption and diarrhoea, with weight loss and anorexia being additional common complaints. Glossitis is a frequent sign of megaloblastic anaemia, with the patient displaying a painful, smooth, red tongue. Other symptoms reported include dyspeptic symptoms, epigastric discomfort, postprandial bloating and fullness, and early satiety.
The elevation in bilirubin levels, caused by ineffective erythropoiesis, manifests as jaundice. Patients may develop neurological symptoms or may be associated with autoimmune diseases such as autoimmune thyroid disease, Type 1 diabetes mellitus, and vitiligo, as part of the autoimmune polyendocrine syndromes.3,4,23,25,26
NEUROLOGIC ABNORMALITIES
Neurologic abnormalities are seen in PA as a result of vitamin B12 deficiency. It could be isolated or be the first manifestation of vitamin deficiency and occur without any haematological or gastrointestinal context. Demyelination is the initial finding, which progresses to axonal degeneration and neuronal death if left untreated. The widely known major manifestations described include peripheral neuropathy, subacute combined degeneration of spinal cord, dementia, ataxia, optic atrophy, psychosis, and mood disturbance. Further neurological disorders described include cerebellar ataxia, abnormalities of cranial nerves, parkinsonism, and movement disorders.26-31
Isolated cases of nominal dysphasia or amnesic aphasia were reported.32,33 A minority of patients exhibit mental or psychiatric disturbances (psychosis) or autonomic signs (bladder, erectile dysfunction, and orthostatic hypotension). Additionally, patients with PA must be closely observed for hypotension and it is also advisable to screen patients with chronic postural hypotension for vitamin B12 deficiency.34 Epilepsy is rarely seen in adult cases. The appearance of motor symptoms is indicative of subacute combined degeneration involving the dorsal and lateral spinal columns. Imaging of the spinal cord in cases of severe myelopathy that are not initially recognised as the result of vitamin B12 deficiency, had characteristic hyperintensity on T2-weighted imaging, described as an inverted V-shaped pattern in the cervical and thoracic spinal cord.28,29 It is particularly important to recognise these symptoms early because the neurological lesions may not be reversed after replacement therapy with vitamin B12.
GASTRIC CANCER AND PERNICIOUS ANAEMIA
Patients with PA may also be at a higher risk for developing gastric cancer (GC) (adenocarcinoma and gastric carcinoid Type I) as an end-stage evolution of atrophic gastritis. Hypergastrinaemia, secondary to hypochlorhydria in PA patients, is a well-known risk factor for enterochromaffin-like cell hyperplasia and gastric carcinoids. Hypochlorhydria leads to overgrowth of nitrosamine producing bacteria with potential carcinogen activity.35 However, more extensive atrophy of the gastric mucosa (multifocal atrophic gastritis) seems to be associated with an increased risk of progression to gastric neoplastic lesions.35
The meta-analysis presented by Vannella et al.36 reported that PA is associated with a nearly 7-fold relative risk of GC and the incidence-rate of GC in PA is 0.27% per person-years. Furthermore, a recent meta-analysis by Lahner et al.37 showed an overall lower relative risk of cancers other than GC in PA patients, but an increased relative risk of biliary tract cancers and haematological malignancies was observed. The increased risk for the development of gastric neuroendocrine tumours in patients with PA represent an additional potential rationale for endoscopic surveillance in these patients.38 The management of patients with PA remains controversial and the need for endoscopic and histological surveillance strategies to prevent GC in these patients is not universally accepted. However, in young patients, and when endoscopy detects any preneoplastic characteristic lesions, most experts agree that it is convenient to perform an endoscopic evaluation at the diagnosis of PA and then every 2–5 years. Additionally, the British Society of Gastroenterology (BSG) guidelines recommended a follow-up with endoscopic surveillance every 3 years to patients with extensive GA. PA patients should be monitored regularly using gastroscopy with antral and corporal biopsies.39
PERNICIOUS ANAEMIA PRESENTING WITH HYPERHOMOCYSTEINEMIA ASSOCIATED THROMBOEMBOLISM
Several case-control studies and even a meta-analysis have confirmed a link between venous thrombosis and hyperhomocysteinemia. Homocysteine is due to genetic and acquired factors (poor diet in folate and vitamin B12, older age, renal impairment, thyroid diseases, and malignancies) induced by the intake and the concentrations of vitamin B9 or B12 in the majority of cases.40,41 However, the most common cause of vitamin B12 deficiency with hyperhomocysteinemia is PA.
It was thought that the main pathophysiological link among these vitamins and venous thrombosis is the accumulation of homocysteine as a result of decreased concentrations of these B group vitamins. However, all of these vitamins have a homocysteine-independent role related to the development of venous thrombosis. In addition, hyperhomocysteinemia inhibits the inactivation of factor Va by activated protein C and could increase the effect of factor V Leiden. Many hypotheses have been suggested to explain how hyperhomocysteinemia may lead to venous thrombosis. One hypothesis is that homocysteinemia has a toxic effect on the vascular endothelium and on the dotting cascade.40 Addittionally, homocysteine has several procoagulant properties including the decrease of antithrombin III binding to endothelial heparan sulfate, increase of affinity between lipoprotein(a) and fibrin, induction of tissue factor activity in endothelial cells, and inhibition of inactivation of factor V by activated protein C.42,43 Understanding the molecular pathogenesis of the development of thrombosis in patients with hyperhomocysteinemia related to Biermer’s disease would help us identify patients at risk and treat them accordingly. Thus, these conditions should remain in the clinician’s mind, especially when thrombosis occurs along with biological abnormalities such as anaemia, megaloblastosis, or haemolysis.44
DIAGNOSIS OF PERNICIOUS ANAEMIA
The diagnosis of PA relies on the presentation of megaloblastic anaemia, low serum vitamin B12 levels, gastric atrophy, and the presence of antibodies to gastric parietal cell or IF (Figure 1).
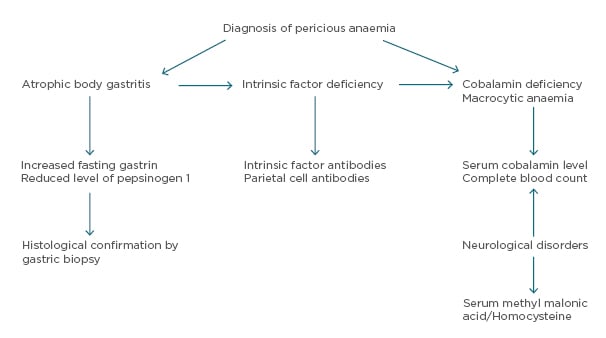
Figure 1: Diagnostic algorithm for pernicious anaemia.8,50
The anaemia is macrocytic and normochromic with a reduction in the absolute number of reticulocytes. The patient’s red blood cells exhibit marked anisopoikilocytosis and numerous oval macrocytes. Although the hypersegmented neutrophils support the diagnosis of megaloblastic anaemia, they are not specific. The variation in the size and shape of red blood cells could lead to a misdiagnosis of microangiopathic haemolytic anaemia instead of megaloblastic anaemia. Some patients may present with nonanaemic macrocytosis for several months before the diagnosis of PA is made. Additionally, macrocytosis is absent in 30% of PA patients if iron deficiency is associated. Moreover, there may be masking of macrocytosis by α-thalassemia in PA patients with African origins who might present with microcytosis. Pancytopenia is often present with rates ranging from 5 to 37%.45
Schistocytes may be seen in megaloblastic anaemias as a reult of erythroblast cytoskeletal fragility, reflecting the severity of dyserythropoiesis. Several cases of PA presenting with pseudothrombotic microangiopathy are reported in the literature and treated with plasma transfusions and exchange. These cases are characterised by haemolysis, thrombocytopenia, and schistocytosis with higher mean LDH levels.46 Very high LDH levels, mild-moderate thrombocytopenia, and a low reticulocyte count are strongly suggestive of pseudothrombotic microangioapthy and should prompt the physician to screen for cobalamin deficiency.47
Bone marrow biopsy and aspiration are not necessary for the diagnosis of megaloblastic anaemia (Figure 2) and may be misleading in cases of severe pancytopenia with hypercellularity, increased erythroblasts, and even cytogenetic abnormalities, confusing the diagnosis with acute leukaemia. It shows a hypercellular bone marrow with a shift toward immaturity and abnormal maturation of erythroid and myeloid cell lines. The immature neutrophil series exhibits nuclear-cytoplasmic asynchrony with numerous giant metamyelocytes. The ineffective erythropoiesis and myelopoiesis are responsible for the pancytopenia in megaloblastic anaemia, despite marrow hypercellularity.3,4
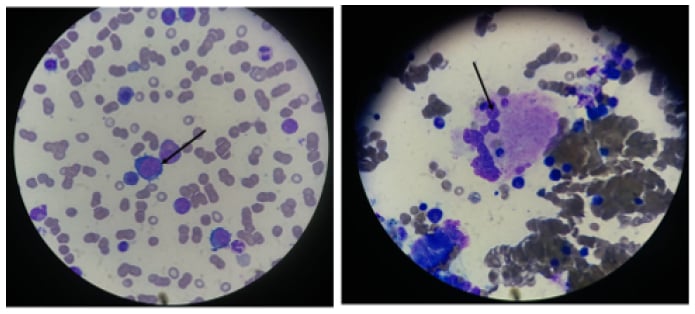
Figure 2: Bone marrow biopsy displaying megablastosis.
A bone marrow aspirate shows megaloblastic features. Large erythroblasts and other red-cell precursors are characterised by an open, immature nuclear chromatin pattern. There is dyssynchrony between the maturation of the cytoplasm and that of the nuclei in later red-cell and granulocyte precursors.
Photographs courtesy of Dr Benkirane.
Serum vitamin B12 and serum folate levels should be determined concurrently to correctly identify patients deficient in either or both; however, there exists limited sensitivity and specificity. Differentiating between vitamin B12 deficiency and folate deficiency is essential to patient management because treatment of vitamin B12 deficient patients with folate alone may reverse the megaloblastic blood picture, although the associated neurologic damage may worsen. Cobalamin level is measured by an automated competitive-binding immunoenzymatic luminescence method, the results of which may not always accurately reflect actual vitamin B12 stores. Low levels (<100 pg/mL) are usually associated with clinical deficiency, however both false-negative and false-positive values are common (occurring in up to 50% of the tests), attributable to the fact that only 20% of the total measured vitamin B12 is on the cellular delivery protein TC; the remainder is bound to haptocorrin, a protein of unknown function. IF antibodies may bind the test IF reagent and if there is a failure in the denaturation step intended to denature IF-blocking antibodies, spuriously normal or increased vitamin B12 levels can be measured.48,49 In this situation, measurement of serum methylmalonic acid and total homocysteine is useful in making the diagnosis of vitamin B12 (markedly elevated levels) deficiency in patients who have not received treatment, including those who have only neurologic manifestations of deficiency.
ELEVATED LEVELS OF TOTAL HOMOCYSTEINE AND METHYLMALONIC ACID
Methylmalonic acids have been proven as markers for insufficient intracellular vitamin B12 because the levels can be remeasured to document adequate vitamin B12 replacement. An elevated level of methylmalonic acid is reasonably specific for vitamin B12 deficiency, and the level always decreases with vitamin B12 therapy. The level of serum total homocysteine is less specific because it is also elevated in folate deficiency, classic homocystinuria, and renal failure.
A deficit of IF may be demonstrated using the Schilling test, a dynamic multistep investigatory test which involves the ingestion of isotope-labelled vitamin B12, followed by an injection of unlabelled vitamin B12. Given its complexity and problems related to the use of radioactive agents, the Schiling test is now being replaced by other diagnostic strategies such as the detection of IFA.
A positive test for anti-IFA or anti-parietal-cell antibodies (by immunoblotting, ELISA, and chemiluminescent immunoassay methods) identifies an autoimmune basis for the gastric atrophy in PA. Anti-parietal-cell antibodies are found in 90% of patients with PA, but have low specificity and are seen in atrophic gastritis without megaloblastic anaemia as well as in various autoimmune disorders. IFA are less sensitive as a result of only being found in 60% of patients with PA, but are considered highly specific for PA. However, a positive correlation between the increasing histological score of body mucosa atrophy and the titer of both antibodies can be observed.18,39 Surveillance for autoimmune thyroid disease is reasonable in patients with positive antibody tests. A diagnostic workup of megaloblastic anaemia should also include evaluation of iron because the bone marrow is overloaded with iron that cannot be utilised during the megaloblastic state. Therefore, iron supplementation may be warranted even though the patient has an initial normal serum iron value.
Chronic atrophic gastritis can be diagnosed on the basis of an elevated fasting serum gastrin level and a low level of serum pepsinogen I. Some experts recommend endoscopy to confirm gastritis and rule out gastric carcinoid and other GC because patients with pernicious anaemia are at increased risk for such cancers. In PA patients, the mucosa of the cardia and fundus is thinned and atrophied, with shrunken glands and containing few principal and parietal cells, while usually the mucosa of the antrum is spared. However, a concomitant antral atrophic gastritis may be observed in 25% of PA patients.50 These data strongly suggest that an extension of gastritis to the gastric antrum does not necessarily exclude the diagnosis of PA and the presence of gastric autoimmunity.
Differential Diagnosis
Accurate differential diagnosis of other causes of macrocytic anaemia and cobalamin deficiency is mandatory (Table 1).

Table 1: Other causes of macrocytic anaemia and cobalamin deficiency.3,4
TREATMENT
The clinical management of patients with PA has two different aspects: the treatment of cobalamin deficiency and the monitoring of iron deficiency onset. PA is caused by inadequate secretion of gastric IF, which is necessary for vitamin B12 absorption and thus cannot be treated with oral vitamin B12 supplements.
The therapeutic recommendations for PA, with regard to dosage and administration of vitamin B12 substitution treatment, are divergent. Vitamin B12 must be administered parenterally and patients generally receive an intramuscular injection of 1,000 µg B12 every day or every other day during the first week of treatment. The next month, they receive injections every week, subsequently followed by monthly injections.
The alternative to intramuscular B12 injection is high-dose oral B12, to which a 1,000–2,000 µg/day dose has been demonstrated to be effective.51 However, despite many studies suggesting oral administration of vitamin B12 to be easy, effective, and less costly than intramuscular administration, debate surrounds the effectiveness of the oral route. Patients should be offered this alternative after an informed discussion on the advantages and disadvantages of both treatment options. The effect of oral cobalamin treatment in patients presenting with severe neurological manifestations has not yet been adequately documented. Although, recommendations are to always use the parenteral route in severe neurological manifestations. Approved sublingual and intranasal formulations of B12 are also available.52,53
PA requires lifelong treatment and the percentage of vitamin B12 absorption improves with supplementation, but symptoms of vitamin B12 deficiency may be improved after just a few days of medical treatment. Gastric atrophy, however, is not cured by cobalamin.
MONITORING
The earliest sign of treatment response is an increase in reticulocyte count, usually within 3 days of treatment. Following changes in the decrease of biochemical markers such as methylmalonic acid and plasma homocysteine levels have been observed in the first 5 days of treatment.4,8 Sustained normalisation of serum cobalamin usually occurs following 2 weeks of therapy.
The macrocytosis correction takes place during the first month of treatment. The surveillance of these patients is mandatory to detect early long-term consequences of PA, such as GC and carcinoids.50 A clinical interview should be considered every year to attest the commencement of new symptoms. These may include epigastric pain, dysphagia, iron deficiency, and/or others that require gastroscopic investigation. The key management principle is the importance of routine follow-up.
CONCLUSION
PA is an underdiagnosed autoimmune disease. It is a complex disorder consisting of haematological, gastric, and immunological alterations. Macrocytic anaemia is the result of vitamin B12 deficiency, which, in turn, is the result of deficiency of IF corpus atrophy. The diagnosis of PA remains challenging in many circumstances for many clinicians because of its diverse clinical manifestations and the limitations of currently available diagnostic tools. Early detection and treatment have led to a lower percentage of vitamin B12 deficiency patients with PA.





