
Abstract
Acute lymphoblastic leukaemia (ALL) in adults has a survival rate of 40–50% at 5 years, with a high relapse rate after first-line chemotherapy. After relapse, results with salvage therapy are currently unsatisfactory. Therefore, both the optimisation of front-line therapy to reduce relapse incidence and the search for effective salvage therapies for relapsed/refractory (r/r) ALL have been of great interest to the medical community in recent years. The well-characterised expression of well-defined cell-surface antigens in B cell ALL (B)-ALL and T cell (T)-ALL, such as CD19, CD20, CD22, and CD52, has led to the development of several immunotherapy strategies, comprising ‘nude’ monoclonal antibodies (moAbs), conjugated moAbs, bispeciphic, or highly sophisticated chimeric antigen receptor (CAR)-T cell therapy.
Recently, both the bispecific moAb blinatumomab (anti-CD19 coupled with a CD3 recognition subunit) and the conjugated anti-CD22 moAb inotuzumab-ozogamicin have resulted in higher remission rates (44% versus 25%, and 80.7% versus 29.4%, respectively) and survival advantages (median overall survival [OS]: 7.7 months versus 4 months, and 7.7 months versus 6.7 months, respectively) in patients with r/r B-ALL when compared to standard salvage chemotherapy-based regimens. On the other hand, preliminary reports show feasibility and unprecedented response rates of ≤90% in highly refractory children and adults treated with CAR-modified T cells targeting the B cell specific CD19 antigen, which seem to be durable in a significant proportion of patients. Furthermore, the addition of anti-CD20 moAb rituximab to front-line standard chemotherapy in patients with CD20+ B-ALL has resulted in a clinical benefit, with prolongation of response duration and survival (3-year leukaemia-free survival and OS: 70% versus 38%; p<0.001, and 75% versus 47%; p=0.003).
In conclusion, immunotherapy is currently providing additional options for high-risk ALL patients both in front-line or advanced phase. Nonetheless, the optimal positioning of these novel agents, specially in relation to allogeneic haematopoietic stem-cell transplantion, needs to be clarified. This article aims to review several of these new therapeutic immunotherapy options available for patients with adult ALL, as well as their specific toxicity profile.
INTRODUCTION
Acute lymphoblastic leukaemia (ALL) is the most common childhood malignancy, with an incidence of 5 per 100,000.1 In adults, ALL is less frequent, with an incidence of ˜1 per 100,000.2 Whereas the 5-year overall survival (OS) in children is ˜90%, outcomes in adults are less favourable, at ˜40%.2,3 In adults, despite an initial high response rate of 80–90% with front-line treatments, a significant proportion of patients experience a relapse. Applying paediatric-like regimens to adolescents and young adults optimises front-line therapy efficiency.4 After a first relapse, the chance to obtain a subsequent complete remission (CR) with standard chemotherapy-based regimens ranges from 30–40%, and decreases to 10–20% after further relapses. Moreover, only a minority of these patients will be able to receive allogeneic haematopoietic stem cell transplantation (allo-SCT); this is the only treatment that allows durable responses in a significant proportion of relapsed ALL patients.5-7
In this context, there is a great interest in optimising front-line therapy to prevent relapse and find more effective salvage therapies for relapsed/refractory (r/r) ALL. The stable expression of several antigenic markers in B cell ALL (B-ALL) and T cell ALL (T-ALL), such as CD19, CD20, and CD22 cell-surface antigens, has led to the development of diverse antibodies with significant clinical activity directed against these antigens. Antibodies such as blinatumomab, inotuzumab-ozogamicin (IO), and rituximab have already been approved by the US Food and Drug Administration (FDA).8-10 Recently, chimeric antigen receptor (CAR)-T cell therapy has shown promising results in treatment of r/r ALL, although it is still in the early stages of development.11,12
CD19-DIRECTED THERAPY
CD19 is a cell-surface receptor expressed on B cells from the late pro-B cell stage until plasma cell differentiation.6,7 Blinatumomab, approved by the FDA (2014) and the European Medicines Agency (EMA) (2015) for the treatment of r/r philadelphia-negative (Ph-) B-ALL.
Blinatumomab is a bispecific T cell engager antibody, derived from a B-linage-specific mouse monoclonal antibody (moAB). Thus, one arm binds CD3 while the other binds CD19, redirecting unstimulated primary human T cells against CD19-positive lymphoma cells.8,13 Activating proliferation of CD4+ and CD8+ cells and inducing granzyme and perforin-mediated serial tumour lysis upon recognition of CD19 antigen in B-cells, including B-ALL blasts.3,5,6,14
Blinatumomab has been administered in two different clinical settings with different tumour loads: in patients with persistent or reappearing minimal residual disease (MRD) and for patients in overt haematological relapse. Interestingly, in a small series of patients with positive MRD, blinatumomab was capable of inducing durable responses with MRD clearance in 16 out of 18 patients.15 Furthermore, this agent showed a high clinical activity in r/r Ph- B-ALL. Thus, the response rate in a Phase II trial that included 189 patients with high-risk relapse was 43%, within two courses, including 33% CR. Forty percent of treated patients subsequently received an allo-SCT. Adverse events included two unexpected, blinatumomab-specific events, such as Grade 3 cytokine release syndrome (CRS) in 3 patients and Grade 3–4 neurological events in 20 patients, which included seizures, aphasia, and encephalopathy. This drug must be administered in continuous intravenous infusion over 4 weeks at a dose of 28 μg/day (9 μg/day during the first week).15,16
In a confirmatory Phase II study of 116 adult patients, 78% had a complete MRD response rate after one cycle of blinatumomab. The most frequent adverse events were tremor, aphasia, and encephalopathy.17 In a Phase II study in r/r Philadelphia-positive B-ALL, 44 of 45 patients failed the second or later-generation tyrosine kinase inhibitor therapy, of which 16 (36%) patients achieved CR or CR with partial haematological recovery. The adverse events were those described previously.18
A recent, large, Phase III study has shown the superiority of blinatumomab compared to standard salvage chemotherapy in patients with refractory or high-risk (i.e. with a response duration <12 months or following allo-SCT) B-ALL r/r in terms of response rate (44% versus 25%) regarding CR and CR with incomplete haematological recovery and survival. Nonetheless, median OS after blinatumomab was 7.7 months, and blinatumomab is recommended as a ‘bridge strategy’ to bring patients to allo-SCT, although the overall result of such a strategy is currently unknown. Although the mechanisms of resistance to blinatumomab are incompletely elucidated, the emergence of a CD19 negative subclone can be observed in 30–50% of patients.19
CAR are recombinant antigen receptors with an anti-tumour target specificity, generated with the purpose to redirect autologous or allogeneic T lymphocytes or natural killer cells against tumour cells.20 Diverse CAR T-cells have been engineered with different antigenic specificity and several costimulatory constructs to induce a durable response. Nonetheless, most clinical experiences accumulated correspond to treatment of advanced phase B-ALL and other B cell malignancies with anti-CD19 CAR-T cells. CD19 is considered an adequate target for treatment of B cell malignancies, since it is a surface marker expressed during all B cell ontogeny and expressed in almost all B cell malignancies, including ALL.20 Adoptive transfer of anti-CD19 CAR-T cells has resulted in an unexpectedly high rate of response in highly refractory B-ALL populations,20,21 with CR rates of ≤90% and 1-year survival >50%, in both children and adults.
Experience with CTL019, anti-CD19 CAR with 4-1BB (CD137) costimulatory molecule (University of Pennsylvania, Pennsylvania, USA) in paediatric and adult B-ALL reported 27 of 30 patients (90%) were in a morphologic CR at the first assessment, 1 month after the infusion of CTL019. The rate of event-free survival at the median follow-up of 6 months was 67% in this heavily pretreated population.20
Davila et al.,11 in a clinical trial involving 16 patients with r/r B-ALL who were treated with autologous T cells expressing the CAR 19-28z specific for the CD19 antigen, demonstrated that the response rate was 88%, allowing the transition of most of these patients to an allo-SCT as definitive therapy.11 Lee et al.22 recently reported a CR rate of 70% in a National Cancer Institute (NCI) analysis of 20 children and young adults with ALL.22,23 Overall, there has been substantial experience with CAR therapy in children and adults with B-ALL, and response rates have not varied with age.
Treatments with CAR-T have been associated with unexpected toxicity, considered to be related to the rapid expansion of CAR-T clones, namely CRS and neurological dysfunction.11 A few cases of CRS and neurotoxicity with fatal outcome have been described. Laboratory markers of systemic inflammation, including C-reactive protein and ferritin levels, were elevated in all the patients. Patients who had severe CRS had higher peak levels of interleukin (IL)-6 than patients who did not have severe CRS (p<0.001). Predictive factors and optimal management (treatment with tocilizumab/anti-IL-6 for severe CRS) resulted in a complete reversal of symptoms and a normalisation of laboratory results. Relapses ocurred in two of the nine patients who received immunosuppressive therapy for the CRS.11,22
Moreover, therapy with anti-CD19 CAR-T is followed by persistent B cell aplasia (on-target toxicity), which requires immunoglobulin reposition.11,12,22,24-26 Randomised controlled trials comparing the efficacy of CD19-CAR versus blinatumomab r/r B-ALL are not available. CAR-based therapies have, on average, demonstrated higher remission rates than those reported with blinatumomab24-26 A major distinction between blinatumomab and CD19-CAR-T cells is a difference in the duration of anti- leukaemic effects (CD19-CAR>blinatumomab).24-27
In summary, CD19-targeted CAR-T therapy is an emerging therapy that results in a high rate of response in both children and adult B-ALL refractory, compared to other therapeutic alternatives; responses that are durable in a significant proportion of patients. Although CAR-T cell therapies are still at an early stage, several observations suggest potential benefits. To date, the most dramatic results have been seen in ALL.11,12,22
A number of issues surrounding this therapy remain uncertain: extended follow-up is required to assess the proportion of patients that can be cured, the requirement of subsequent allo-SCT, optimal anti-CD19 CAR-T construct, the management of toxicity, predictive factors of response, and overall further development (CAR-T with dual targets, etc.). The worldwide availability of this therapy is also unclear.
ANTI-CD20 THERAPY
Rituximab is a chimeric moAb against CD20. Patients with B-lineage ALL may also have the CD20 antigen, being one of the first moAb that was evaluated as a treatment for patients with ALL.28 Interest in this kind of condition is because ˜30–50% of precursor B cells express the CD20 antigen on the surface, which is targeted by rituximab. In addition, there are data suggesting that the expression of CD20 carries a worse prognosis in ALL.29
Maury et al.,10 in a multicentre trial with adults with CD20-positive (CD20+) Ph- ALL randomised patients to receive chemotherapy with or without rituximab and demonstrated that patients assigned to the rituximab group had longer event-free survival than those assigned to the control group with GRAALL-2005 chemotherapy regimen (hazard ratio [HR]: 0.66; 95% confidence interval [CI]: 0.45–0.98; p=0.04). The estimated 2-year event-free survival rates were 65% (95% CI: 56–75) and 52% (95% CI: 43–63), respectively. The safety profile was quite good and the study showed that infectious events were slightly more frequent in the rituximab group, but the difference was not significant.
Similarly, Thomas et al.30 in a prospective, sequential, open-label, single-centre, Phase II trial, treated 282 adolescents and adults with newly diagnosed Ph- B-ALL with standard or modified hyper-CVAD regimens. The latter incorporated standard dose of rituximab when the expression of CD20 was >20%. The results suggested that the addition of rituximab to the hyper-CVAD-based regimens for the CD20+ precursor ALL-B significantly improved CR rates at 3 years (70% versus 38%; p<0.001) and OS (75% versus 47%; p=0.003).30
The German group was able to demonstrate improvement in the 3-year CR and OS rates (64% versus 58%; p=0.009; and 75% versus 54%, no p value given) when incorporating rituximab to standard chemotherapy in the context of the GMALL protocol 07/2003.31,32 Studies to date have demonstrated the advantages of the association of rituximab in standard therapeutic protocols for patients with CD20+ Ph- ALL, with a safety profile quite similar to standard therapy.10,33
Recently, second-generation anti-CD20 moAbs, such as ofatumumab (Type I) and obinutuzumab (Type II), have been used in CD20+ B-ALL. Addition of ofatumumab to hyper-CVAD chemotherapy has been used in a Phase II trial with a limited number of patients with promising results, and is currently being used in an ongoing clinical trial in combination with BFM chemotherapy.34-36
CD22-DIRECTED THERAPY
CD22 is a B-lineage specific antigen expressed in >90% of leukaemic blasts in patients with ALL. It is a 135 kDa sialoglycoprotein that is expressed from the early to late stages of differentiation of B cells, with loss of expression in plasma cells.37 CD22 is part of the immunoglobulin superfamily and what is interesting, from a pharmacological point of view, is that CD22 is rapidly internalised when binding to its antigen occurs, so it can be conjugated to a cytotoxic component to increase its efficacy (Figure 1).38
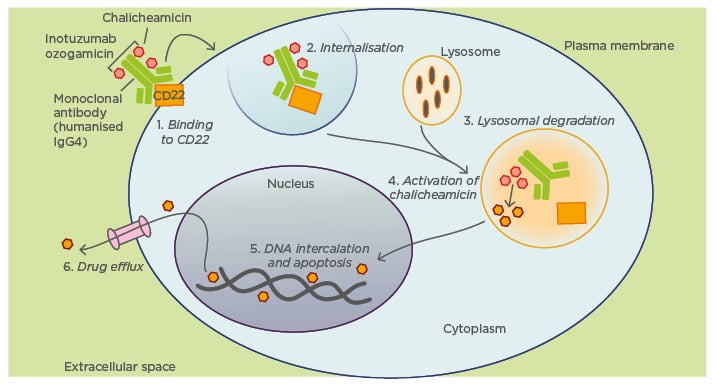
Figure 1: Proposed mechanism of action of inotuzumab ozogamicin.
IgG4: immunoglobulin G4.
IO is a humanised moAb against CD22 conjugated to calicheamicin, a cytotoxic agent derived from Micromonospora echinospora.39 Calicheamicin induces breaks in DNA double-strands and apoptosis independent of cell cycle progression.40 IO is the most developed targeted antibody in this group. IO’s ability to induce cell death is dose and time dependent and maximal saturation of CD22 is not essential for efficient cell death. The hypothesis is that there is a continuous renewal of the cell surface expression of CD22 with an unstopped binding of IO and subsequent accumulation of calicheamicin inside the cell, which leads to apoptosis.31
A two-group, randomised, Phase III trial, assigned adult patients with CD22+ r/r ALL to receive either IO or standard intensive chemotherapy.42 IO was given intravenously at a 1.8 mg/m2 dose receiving 0.8 mg on Day 1 and 0.5 mg on Days 8 and 15, based on a Phase II clinical trial. The first cycle had a duration of 21 days and from the second cycle the duration was 28 days. The other group received chemotherapy of investigator’s choice with three possible standard regimens.
Of the 326 patients who underwent randomisation, the first 218 (109 in each group) were included in the primary intention-to-treat analysis of complete remission. The primary endpoints were CR and OS. The results showed a CR of 80.7% versus 29.4% (p<0.001) and OS of 7.7 months versus 6.7 months (HR: 0.77; p=0.04). High remission rates were noticed in patients with both higher (>90%) and lower (<90%) levels of CD22 expression. Other results showed a CR with MRD below the threshold (0.01% marrow blasts) of 78.4% versus 28.1% (p<0.001), duration of remission of 4.6 months versus 3.1 months (HR: 0.55; p=0.03), and progression free survival 5 months versus 1.8 months (HR: 0.45; p<0.001). More patients proceeded to SCT after treatment in the IO group (41% versus 11%; p<0.001). It is noticeable that in patients with Ph+ ALL or t(4:11)+ ALL remission rates did not differ significantly between treatment groups.
Liver-related adverse events were more frequent in the IO group. They included increased levels of aspartate aminotransferase, alanine aminotransferase, and bilirubin. Veno-occlusive liver disease (VOD) of any grade occurred in 15 patients (11%) and cases were reported ≤2 years after randomisation. During, or shortly after treatment, five patients were diagnosed with VOD, two of which had undergone a SCT before randomisation. Of the 45 patients of the IO group who underwent a SCT after the trial, 10 patients had VOD, with the procedure being the second transplantation for 3 patients. The median time to the development of VOD after SCT was 16 days. Two treatment-related deaths due to VOD occurred after post-trial transplantation. In the standard chemotherapy group no cases of VOD were noticed during treatment and, of the 20 patients who underwent SCT, 1 patient was diagnosed with VOD.42,43
A study to identify prognostic factors in r/r ALL patients receiving IO was performed. By multivariate analysis, a high peripheral blood absolute blast count (>1×109/L) and low platelet count (<100×109/L) was associated with a lower chance to achieve bone marrow CR. Also, baseline features were seen to independently affect survival, including cytogenetics (complex karyotype, translocation [4;11], translocation [9;22], abnormal chromosome 7), disease beyond first salvage therapy, and high peripheral absolute blast count. Based on the three previous features, patients with 0, 1, 2, or 3 adverse factors had a median survival of ≥42, 8.8, 7.1, and 2.6 months, respectively.42
IO is also being tested as a first-line treatment in old patients given the high toxicity related to conventional chemotherapy. There is an ongoing Phase II clinical trial for patients ≥60 years of age with newly diagnosed ALL who receive a combination of low intensity chemotherapy with mini-hyper-CVD, IO, rituximab, and intrathecal chemotherapy.38 Of the 20 patients included, with a median age of 69 years, an overall response rate of 95% has been observed, with 75% CR and 20% C-reactive protein. Progression free survival at 1-year has been 83% and OS 84%. Grade 3–4 non-haematological toxicities included increased liver enzymes and VOD in 1 patient; these are similar adverse effects to those observed in previous studies. These results suggest that combination of low intensity chemotherapy to targeted antibodies in first-line treatment of the elderly may obtain better outcomes than standard therapy.44
CONCLUSIONS
The development of immunotherapy will provide additional opportunities of treatment in r/r ALL and other haematological diseases. Rituximab combined with conventional chemotherapy improves the results in survival, and recurrence-free survival. CAR-T cell therapy against CD19 has obtained response rates ≤90% in some series. Blinatumomab and IO have achieved better response rates compared to conventional chemotherapy. Safety in these new drugs has been tested and adverse events are acceptable.
Certainly, much remains to be explored in the field of immunotherapy for patients with ALL. Use of these drugs as treatment is needed to verify results obtained in clinical trials and further studies are needed to examine long-term results. The next steps to be taken should be exploring the results of immunotherapy as first-line treatments (combined with standard chemotherapy or not) and finding new drugs to improve outcomes in T-ALL.





