
Abstract
Chronic neutrophilic leukaemia (CNL), chronic eosinophilic leukaemia-not otherwise specified (CEL-NOS), and myeloproliferative neoplasm (MPN), unclassifiable are rare clonal diseases, known as ‘non-classic myeloproliferative neoplasms’. They are diagnosed largely based on exclusion of underlying reactive causes by patient history, physical examination, serological tests, and imaging studies. As well as peripheral blood testing, bone marrow examination is mandatory to exclude bone marrow infiltrating conditions such as multiple myeloma, acute leukaemias, etc. Today, molecular genetic classification should be undertaken to establish accurate diagnosis, in addition to the traditional morphological classification of MPN. Therefore, molecular genetic testing should take part in the diagnostic work-up of suspected patients with rare MPN. Of CNL patients, 90% (and in some datasets 100%) have mutations in CSF3R, which has led to the addition of this finding to the diagnostic criteria for CNL. The absence of rearrangements of FIP1L1/PDGFRA, PDGFRA, PDGFRB, FGFR1, and PCM1-JAK2 fusions should prompt consideration of a diagnosis of chronic eosinophilic leukaemia-not otherwise specified. MPN, unclassifiable, the least frequent type, is considered when an MPN has definite MPN features but does not meet diagnostic criteria for either the classic or the other non-classic MPN. They all share common symptoms and findings. Transformation to acute leukaemia is still a major clinical problem. Since no standard of care exists, the treatment approach is still symptomatic for all. This is an indicator that we really need disease-modifying drugs against initial diagnostic molecular markers, such as CSF3R inhibitors, which might change the natural history of these disorders. Therefore, participation in clinical trials is mandatory for this extremely rare patient population.
INTRODUCTION
Myeloproliferative neoplasms (MPN) are rare clonal haematologic diseases characterised by the proliferation of mature blood elements from several myeloid cell lines. The more frequently encountered chronic MPN include polycythaemia vera, essential thrombocythaemia (ET), chronic myeloid leukaemia (CML), and primary myelofibrosis (PMF), respectively. It is important to emphasise that an accurate diagnosis is mandatory for management of and in predicting prognosis in this group of diseases. Today, the traditional morphological classification of MPN is replaced by molecular genetic classification. The main driver mutations in MPN include Janus Kinase 2 (JAK2), calreticulin (CALR), and myeloproliferative leukaemia virus oncogene (MPL). The presence of a mutation associated with an MPN subtype is specific for diagnosis; however, the absence of JAK2, MPL, or other related mutations is not sufficient to exclude the diagnosis of MPN. Common findings of MPN include organomegalia, increased metabolism related constitutional symptoms, and hypercellular bone marrow (BM) with blast percentage <20%. The primary treatment goals in MPN are to avoid thrombosis and bleeding, treat MPN-related symptoms, improve quality of life, and minimise the risk of malignant transformation and myelofibrosis. Leukaemic transformation is associated with adverse clinical outcomes, characterised by a poor response and early resistance to conventional therapies. Current MPN clinical research is focussed on the development of prognostic biomarkers and development of drugs that can modify disease natural history.
In 2016, the World Health Organization (WHO) revised the classification of MPN (Table 1).1 The most seen classical MPN (polycythaemia vera, ET, CML, and PMF) are beyond the scope of this review, and will not be discussed.2-4 Namely, the less frequently encountered ones (chronic neutrophilic leukaemia [CNL], chronic eosinophilic leukaemia-not otherwise specified [CEL-NOS], and myeloproliferative neoplasm, unclassifiable [MPN-U]) will be reviewed extensively.

Table 1: World Health Organization (WHO) 2016 classification of myeloproliferative neoplasms.1
CHRONIC NEUTROPHILIC LEUKAEMIA
CNL is a rare MPN, of which approximately 150 cases have been described to date; however, only 40 ‘true’ cases meet the criteria of the last WHO classification.5 The median age is 65 years (26–83 years) at diagnosis, with male predominance.6 The clinical course of CNL is heterogeneous. However, one can state that the disease can be subdivided as chronic phase, accelerated phase, and blast phase, similar to CML.6 An accelerated phase can be considered if progressive neutrophilia, progressive splenomegaly, or worsening thrombocytopenia develop with resistance to previously effective therapy. A blast phase can be considered if BM blast percentage is >20%. Blastic transformation (mostly reported as myeloid) occurs in a significant proportion (˜20%) at a median of 21 months from diagnosis.7 Other definitions that do not fit the accelerated phase and blast phase can be considered as chronic phase.
The majority of patients are asymptomatic at diagnosis, and leukocytosis is detected incidentally in routine laboratory tests. Fatigue is the most common presenting symptom. Other symptoms reported include weight loss, easy bruising, bone pain, and night sweats. The most frequent finding is splenomegaly.7 Splenomegaly may range from mild to massive, and may be related to weight loss, early satiety, and abdominal fullness. Hepatomegaly and lymphadenopathy are uncommon at presentation.8 Skin findings are rare, but may be presented as leukaemia cutis, Sweet’s syndrome, and pyoderma gangrenosum.
The principal feature of CNL is neutrophilic leukocytosis that is characterised by toxic granulation and Döhle bodies in the absence of prominent immature granulocytosis and disgranulopoiesis. Many patients tolerate very high leukocyte counts without having any symptoms or direct evidence of end organ damage. Varying degrees of anaemia and thrombocytopenia are present. Bleeding diatheses are common in CNL due to acquired von Willebrand’s disease and other acquired coagulation factor or platelet function defects. Leukocyte alkaline phosphatase and vitamin B12 levels may be normal or elevated. BM aspirates and biopsies show a myeloid hyperplasia (>90% cellularity) with an increased myeloid:erythroid ratio, which may reach >20:1, and myeloblasts represent <5% of the cells. Erythropoiesis and megakaryopoiesis are typically normal, and dyshaematopoiesis is not present in any cell lineage. Reticulin fibrosis is not significantly increased.
It is diagnosed largely based on exclusion of underlying causes of reactive neutrophilia and/or the lack of specific molecular markers of other haematological malignancies.9 The diagnostic criteria for CNL are displayed in Table 2. Incorporation of molecular studies is helpful to distinguish between benign versus malignant causes of leukocytosis. Colony stimulating factor 3 receptor (CSF3R) is a trans-membrane protein which plays a prominent role in the growth and differentiation of granulocytes.9 More recently, it was observed that CSF3R is recurrently mutated in patients with CNL,10 which has led to the addition of this finding to the diagnostic criteria for CNL in the most recent 2016 WHO classification of MPN.1 The most common acquired mutation in CSF3R is an activating point mutation in the membrane proximal region (CSF3R-T618I) which activates JAK/STAT signalling, so is an oncogenic driver. Ninety percent (in some datasets 100%) of CNL patients have mutations in CSF3R.7 Other much less common activating point mutations include T615A and T640N.10 CSF3R mutations could be determined by molecular techniques, such as next-generation sequencing of DNA from the BM aspirate and peripheral blood. CSF3R is known to signal downstream through both JAK and SRC tyrosine kinase pathways.
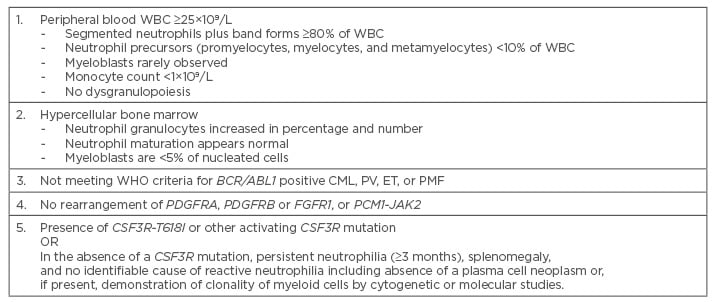
Table 2: Diagnostic criteria for chronic neutrophilic leukaemia.1
CML: chronic myeloid leukaemia; ET: essential thrombocythaemia; PMF: primary myelofibrosis;
PV: polycythaemia vera; WBC: white blood cells; WHO: World Health Organization.
Having a documented CSF3R mutation may help to establish a diagnosis of CNL, but the absence of a CSF3R mutation does not rule out CNL. However, CSF3R-T618I is a highly sensitive and specific molecular marker for CNL. Also, mutations in SETBP1 have been demonstrated in 25% of CNL cases.11 Elliott et al.12 showed that 57% of patients experienced ASXL1 mutations. In another study, Elliott and Tefferi5 concluded that the presence of ASXL1 mutations and thrombocytopenia at diagnosis were independently predictive of shortened survival. Additionally, CNL may occur with monoclonal gammopathy of undetermined significance in ˜33% of cases.6 In cases where the BM shows a plasma cell dyscrasia, it is important to prove the neutrophilic clonality by cytogenetic and/or molecular tests.
CNL should be distinguished from atypical CML (aCML). The diagnosis of aCML is strictly based on morphologic evaluation of the blood and marrow, and there are no specific recurring cytogenetic findings that define aCML. However, the frequency of CSF3R-T618I mutations in aCML ranges from 0–40%.11 Neutrophil precursors are >10% of white blood cells, and monocytosis may be seen in aCML. Another important defining feature in aCML is prominent granulocytic dysplasia. So, a BM evaluation and cytogenetic analysis in suspected cases are required to distinguish these disorders.
CNL is associated with a poor prognosis without established standards of care. Therapeutic options include hydroxyurea, JAK1/2 inhibitor (ruxolitinib), imatinib, or dasatinib, interferon-alpha (IFN-α), hypomethylating agents, thalidomide/lenalidomide, histone deacetylase inhibitors, and chemotherapeutic agents (cytarabine and daunorubicin) with very limited data on therapeutic responses. These agents may improve blood counts, but exhibit no proven disease-modifying benefit. Hydroxyurea is the most commonly used drug and is effective in controlling leukocytosis and splenomegaly.7 Any response to subsequent therapy for hydroxyurea resistance or refractory disease with various agents is often short-lived.7 No haematologic complete remission has been reported to date following standard induction therapy for accelerated or blast phase in CNL. Splenectomy is not recommended for management of disease, because it has a high perioperative mortality rate and has been associated with accelerated neutrophilia. Myeloablative or reduced intensity conditioning regimens followed by allogeneic stem cell transplantation (ASCT) is the only curative option for a minority of patients, because of the rarity of this disease, and the older age of many affected. It has been shown that CSF3R truncation mutations (Y752X, D771fs, S783fs) operate predominantly through SRC kinases, and exhibit drug sensitivity to SRC kinase inhibitors, such as dasatinib, whereas, CSF3R membrane proximal mutations (T618I, T615A) strongly activate the JAK/STAT pathways and are sensitive to JAK kinase inhibitors, such as ruxolitinib.13-15 I suggest using hydroxyurea to control leukocytosis and for evaluation of eligible patients for ASCT. ASCT may result in favourable long-term outcomes in selected patients, particularly when undertaken in the chronic phase of disease, because those who received it in the accelerated phase died after the procedure.16-18 Common causes of death include intracerebral haemorrhage, progressive BM failure and related complications, blastic transformation, and progressive multi-organ failure. Median time to acute myeloid leukaemia (AML) transformation is 21 months (3–94 months) and median survival is 23.5 months (1–106 months).7
CHRONIC EOSINOPHILIC LEUKAEMIA-NOT OTHERWISE SPECIFIED
CEL-NOS is an extremely rare disorder which is due to clonal proliferation of eosinophil precursors. Its true incidence is unknown. The upper limit of normal for absolute eosinophil count (AEC) is ˜600/mm3.5 The severity of eosinophilia can be divided into mild (AEC from the upper limit of normal to 1.500/mm3), moderate (AEC 1.500–5.000/mm3), and severe (AEC >5.000/mm3).19
The most common presenting symptoms are weakness, fatigue, weight loss, night sweats, persistent cough, dyspnea, pruritus, myalgias, rash, fever, rhinitis, and diarrhoea. Patients with CEL-NOS are predominantly male, and splenomegaly is the most frequent clinical manifestation. Other haematologic findings include neutrophilia, basophilia, myeloid immaturity, and both mature and immature eosinophils with varying degrees of dysplasia in peripheral blood or BM.
In general, all organ systems may be susceptible to the effects of sustained eosinophilia. Progressive heart failure is the most important eosinophil-mediated organ injury. Eosinophil infiltration of cardiac tissue and release of toxic mediators from eosinophils lead to endocardial damage with resulting mural thrombi and increased embolic risk. In the later fibrotic stage, fibrous thickening of the endocardial lining causes a restrictive cardiomyopathy.20 Valvular insufficiency results from mural endocardial thrombosis and fibrosis involving leaflets of the mitral or tricuspid valves.21 Besides the heart, lungs, gastrointestinal system, central nervous system, and skin, any other organ may be affected by eosinophil-mediated cytokines.
The diagnostic work-up should begin with eliminating the secondary aetiologies. Secondary eosinophilia has numerous causes. Infections, particularly tissue-invasive parasites, are the most common underlying cause of eosinophilia.21 The patient’s travel history, repeated parasite testing, stool culture, and antibody testing for specific parasites are mandatory for identifying infectious aetiologies. Other potential causes include allergic and hypersensitivity conditions, drug reactions, collagen-vascular diseases (e.g. Churg-Strauss syndrome, systemic lupus erythematosus), pulmonary eosinophilic diseases (e.g. allergic bronchopulmonary aspergillosis), and metabolic conditions (e.g. renal deficiency).22-24 Also, solid tumours such as T-cell lymphomas, Hodgkin’s disease, and acute lymphoblastic leukaemias may be associated with secondary eosinophilia by the production of cytokines such as interleukin (IL)-3, IL-5, and GM-CSF, which promote eosinophil differentiation and survival.25-27 Therefore, chest X-ray, electrocardiogram and echocardiography, computed tomography scan of the chest/abdomen/pelvis, pulmonary function tests, bronchoscopy, and serologic tests (e.g. aspergillus immunoglobulin E) may be additional laboratory investigations. The diagnostic criteria for CEL-NOS are displayed in Table 3.28
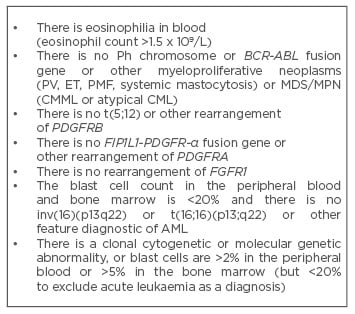
Table 3: Diagnostic criteria for CEL-NOS.28
AML: acute myeloid leukaemia; CEL-NOS: chronic eosinophilic leukaemia-not otherwise specified; CML: chronic myeloid leukaemia; CMML: chronic myelomonocytic leukaemia; ET: essential thrombocythaemia; FGFR1: fibroblast growth factor receptor 1; MDS/MPN: myelodysplastic/ myeloproliferative neoplasms; PDGFRA: platelet-derived growth factor alpha; PDGFRB: platelet-derived growth factor beta; PMF: primary myelofibrosis; PV: polycythaemia vera.
If secondary causes of eosinophilia are excluded, one should proceed to the evaluation of a primary BM disorder. Examination of the blood smear and blood tests (e.g. circulating blasts, dysplastic cells, monocytosis, serum B12, or tryptase level) in conjunction with BM analysis will help to discriminate systemic mastocytosis, CML, AML, myelodysplastic syndrome (MDS), aCML, or chronic myelomonocytic leukaemia. In this context, laboratory evaluation of primary eosinophilia should begin with screening of the peripheral blood for the FIP1L1-PDGFRA gene fusion by reverse transcription polymerase chain reaction (RT-PCR) or fluorescent in situ hybridisation (FISH). FIP1L1/PDGFRA rearrangement is the most common molecular abnormality (range, 3–56%) in chronic eosinophilic leukaemia (CEL).29 We should keep in mind that CEL, CEL-NOS, and idiopathic hypereosinophilic syndrome (HES) are not the same eosinophilic disorders. Formerly, WHO 2008 incorporated CEL/HES into HES under the MPN category, and also described ‘CEL, not otherwise categorised’ (CEL-NOC) and ‘myeloid neoplasms associated with eosinophilia and abnormalities of PDGFRA, PDGFRB and FGFR1’.30 In short, the latter group has been assigned a new category of its own, whereas both HES and CEL-NOC have remained subcategories of MPN according to WHO 2008.30 WHO 2016 revised CEL-NOC as CEL-NOS, and HES and CEL could no longer be assigned as an MPN category. Moreover, many patients, who have been classified as idiopathic HES, can now be found to have a FIP1L1-PDGFRA fusion transcript.31
Briefly, CEL and CEL-NOS are clonal diseases, whilst HES is not. CEL-NOS can be distinguished from CEL by the absence of FIP1L1/PDGFRA rearrangement. CEL-NOS can be distinguished from HES by the presence of a non-specific clonal cytogenetic abnormality or increased blast cells (>2% in the peripheral blood or >5% in the BM, but <20% blasts in both peripheral blood and BM). Furthermore, if FIP1L1-PDGFRA screening is not available, evaluation of the serum tryptase can be a useful surrogate marker for FIP1L1-PDGFRA-positive disease.32 In the absence of FIP1L1-PDGFRA-positive disease, the cases of myeloid and lymphoid neoplasms with abnormalities of PDGFRA, PDGFRB, FGFR1, and PCM1-JAK2 fusions should be excluded. Finally, absence of rearrangements of FIP1L1/PDGFRA, PDGFRA, PDGFRB, and FGFR1, and absence of PCM1-JAK2 fusions should prompt consideration of a diagnosis of CEL-NOS when there is cytogenetic and/or morphologic evidence of an eosinophilic myeloid malignancy that is otherwise not classifiable.33
Cases with CEL-NOS are characterised by aggressive clinical course, and they are usually unresponsive to conventional chemotherapy.34 The efficacy of currently used therapeutic agents is limited, and the haematological responses are usually short-lived. Similar to other MPN, hydroxyurea (1,000 mg/day starting dose) can be used to control leukocytosis and eosinophilia, but with no proven role in favourably altering the natural history of CEL-NOS. IFN-α (starting dose of 1 million units by subcutaneous injection three-times weekly) can produce haematologic and cytogenetic remissions in HES and CEL-NOS patients. The optimal dose and duration of hydroxyurea and IFN-α therapy is unknown and can be tailored to individual response and tolerability. Iurlo et al.35 demonstrated the presence of a KITM541L allele variant in four out of five CEL-NOS patients who were negative for PDGFRA and PDGFRB abnormalities. They concluded that imatinib, which is effective in FIP1L1-PDGFRA rearrangement positive HES, was highly effective also in KITM541L variant cases.35 Namely, imatinib may be considered for some selected CEL-NOS patients. Essentially, imatinib is recommended as a first-line therapy in patients with FIP1L1/PDGFRA-positive CEL.36-38 100 mg/day is sufficient to induce a complete molecular response in most patients with an FIP1L1/PDGFRA fusion gene. Nevertheless, the long-term prognosis, drug withdrawal, and drug resistance therapy remain unclear. Acquired resistance to imatinib can occur because of point mutations in the ATP binding site (e.g. T674I and D842V).39,40 T674I FIP1L1/PDGFRA mutations may be considered as analogous to the T315I mutation in BCR/ABL1, and the prognosis is poor for CEL patients harbouring T674I FIP1L1/PDGFRA. Recently, Jin et al.41 have demonstrated that ponatinib is a potent inhibitor of CEL cells bearing wild-type or T674I FIP1L1/PDGFRA, that induces apoptosis in FIP1LI/PDGFRA expressing cells, making it a promising therapeutic prospect for the future.
The prognosis of CEL-NOS is poor. In a recently reported cohort of 10 patients, the median survival was 22.2 months, and 50% of the patients developed acute transformation to AML after a median of 20 months from diagnosis.42 Three of the five patients who did not develop AML died with active disease; one patient underwent an ASCT and maintained a long-term remission, and the remaining patient achieved a complete remission on imatinib and hydroxyurea.42 ASCT remains a curative option only for younger and fit patients with CEL-NOS.
MYELOPROLIFERATIVE NEOPLASM, UNCLASSIFIABLE
MPN-U is the least frequent subtype of MPN, and little is known about the incidence, clinical features, and management of this rare entity. MPN-U is considered when an MPN has definite MPN features; however, it does not meet diagnostic criteria for either the classic or the other non-classic MPN, or has features that overlap two or more of the MPN categories.43
In a study, CALR mutations were most frequent in patients with MPN-U (37.5%) compared with mutations in the PMF and ET groups (14.8% and 17.7%, respectively).44 Most current data have also indicated that the CALR-mutated group may have a favourable prognosis compared with patients with JAK2 mutations.45,46 A previous study found that CALR mutations were associated with younger age, higher platelet count, lower thrombosis risk, and less leukocytosis, and were less likely to be transfusion dependent.45
MPN-U requires the absence of BCR/ABL1, dyserythropoiesis, disgranulopoiesis, or monocytosis, and the presence of effective clonal myeloproliferation leading to peripheral blood granulocytosis, thrombocytosis, or erythrocytosis. The presence of any one of dyserythropoiesis, disgranulopoiesis, or monocytosis mandates disease assignment to either the MDS or MDS/MPN category. The MDS/MPN are a unique group of myeloid malignancies characterised by both myelodysplastic and myeloproliferative features. MDS-like features include cytopenias and dysplasia of various cell lines while MPN-like features include constitutional symptoms, such as night sweats and weight loss, and elevated blood counts, as well as extramedullary infiltration.47 Therefore, MPN-U should be distunguished from MDS/MPN-U.
In a recent study which is rather indicative of the general characteristics of the disease, 71 consecutive patients with a diagnosis of MPN-U were investigated.48 They found that most of the cases displayed a hypercellular BM (70%) with normal erythropoiesis without left-shifting (59%), increased granulopoiesis with left-shifting (73%), and increased megakaryocytes with loose clustering (96%).48 In this study, megakaryocytes displayed frequent giant forms with hyperlobulated or bulbous nuclei and/or other maturation defects; more than half of the cases displayed severe BM fibrosis (59%); median values of haemoglobin level and white blood cell counts were all within the normal range; in contrast, median platelet count and lactate dehydrogenase were increased. Of the patients, 44% showed splenomegaly. JAK2 mutations were detected in 72% of all patients, and among the JAK2 negative cases, MPL and CALR mutations were found in 17% and 67% of the cases, respectively.48
Currently used therapeutic agents for MPN-U are very limited, and a lack of data prevent us from making a clear recommendation. The very limited case reports from the literature demonstrate hydroxyurea improves blood counts and constitutional symptoms. In addition, the role of medications (except hydroxyurea and transplantation therapy) are not well established. We hope that the accumulating data will help us to understand the whole nature of this rare entity in the near future.
CONCLUSION
In conclusion, the non-classic MPN are rare clonal haematologic diseases, and an accurate diagnosis is mandatory for the management and predicting prognosis in this group of diseases. In this context, molecular genetic testing is the most important tool for establishing diagnosis. However, keep in mind that having a documented disease-related mutation may help to establish an MPN diagnosis, but the absence of a specific mutation does not rule out MPN. The outcomes of current therapeutic approaches, as well as ASCT, are not satisfactory, and also there is no standard of care for CNL, CEL-NOS, and MPN-U. However, great efforts are being spent on discovery of new molecular pathways and development of molecularly targeted therapy. Therefore, participation in clinical trials likely to be conducted is especially encouraged for these extremely rare patient populations.





