
Abstract
Sickle cell disease (SCD) is the most common haemoglobinopathy worldwide and its frequency has steadily increased in Europe in the past decades. SCD is a complex multisystem disorder characterised by chronic haemolytic anaemia, vaso-occlusive crisis, and vasculopathy. Clinical manifestations can be very different, ranging from mild haemolysis to life-threatening acute clinical complications and chronic disabilities. This review will explore service delivery across Europe to children with SCD, reporting on the available minimum standards of care and future perspectives.
INTRODUCTION
Sickle cell disease (SCD) is one of the most common severe monogenic disorders worldwide, with an average of 300,000 children born annually with sickle syndromes.1 SCD was initially endemic in areas with prevalent malarial disease (Africa, the Mediterranean, Southern Asia). In Europe, it was present only in Greece and Southern Italy,2 but historic and recent migration movements have increased the frequency of SCD in areas where it was previously uncommon. In Europe, SCD has become the paradigm of immigration haematology.3 Although considered a rare disease because of its lower frequency in the 28 countries of the European Union (EU), SCD is the most prevalent genetic disease in France4 and the UK,5 and its frequency is steadily rising in many other countries in Northern, Central, and Southern Europe.3,6-10 The World Health Organization (WHO) and the United Nations (UN) recognised inherited haemoglobin (Hb) disorders as a global public health problem in 200611 and 2008,12 respectively, and to face this issue the WHO called upon national health systems “to design, implement, and reinforce …comprehensive national integrated programs for the prevention and management of SCD.”11 Following this invitation, several countries havedeveloped a specific response to meet the needs of patients with SCD, but the recent migrant flow from the Middle East and Africa has further increased the number of patients with SCD in many European countries, subsequently increasing the urgency to fill the gaps in specialised healthcare delivery to this vulnerable group of patients.
SCD is a chronic and complex multisystem disorder requiring comprehensive care that includes screening, prevention, health education, and management of acute and chronic complications.13,14 Poor service organisation and episodic healthcare lead to higher rates of acute events and chronic complications with increased burden on hospital structures and higher costs for health systems.15
The European Hematology Association (EHA) recently published a consensus document, a ‘Roadmap for Hematology Research in Europe’16 with the aim of describing the major achievements in diagnosis and treatment of blood disorders in Europe and identifying the greatest unmet clinical and scientific needs. For SCD, the necessity to improve treatment strategies for both acute and chronic complications was identified as a key area of intervention for optimising patient care, in order to positively affect patient health and quality of life, and to reduce hospitalisation length, patient disability, and cost of care.
This review will assess the management of children with SCD in European countries by exploring the current situation in the delivery of minimum standards of care, health service availability, and future perspectives.
SICKLE CELL DISEASE CLASSIFICATION AND CLINICAL MANIFESTATIONS
The term SCD is used to describe all the different genotypes that cause the characteristic clinical syndrome.13 Its most frequent variant (sickle cell anaemia or homozygous SS disease) is caused by a single amino acid substitution at the sixth residue of the β-globin subunit (p.Glu6Val), which results in the production of the characteristic sickle Hb. Several double heterozygous forms also give rise to the clinical picture of SCD, the most common being the double heterozygous Sβ-thalassaemia° and SCD. The double heterozygous Sβ-thalassaemia° (S mutation coupled with a thalassaemia β° mutation) is the most severe form with a clinical picture similar to homozygous SS disease, while the double heterozygous SCD (in which the Hb composition is approximately 50% HbS and 50% HbC) and the Sβ-thalassaemia+ (a condition that presents a minimal amount of HbA) display intermediate severity. The genetic variants leading to SCD, including the less common ones, are shown in Table 1.

Table 1: Genetic variants leading to sickle cell disease.
The mutated Hb leads to changes in the shape and behaviour of red blood cells resulting in haemolytic anaemia, vaso-occlusion, and vasculopathy, which are the hallmarks of SCD pathophysiology. Other factors such as hypercoagulability, inflammation, and hypoxia-reperfusion are also involved in the organ damage of SCD.
Despite being a monogenic disorder, SCD presents with extreme phenotypic variability. Some patients remain virtually asymptomatic, while others suffer repeated acute events requiring hospital admissions and chronic organ damage with increasing disabilities. The reasons for this variability have not been completely clarified, although a percentage of HbF and co-inheritance of α-thalassaemia and glucose 6-phosphate deficiency can have a role.13,17-19 The most common clinical manifestations occurring in children with SCD are shown in Table 2.
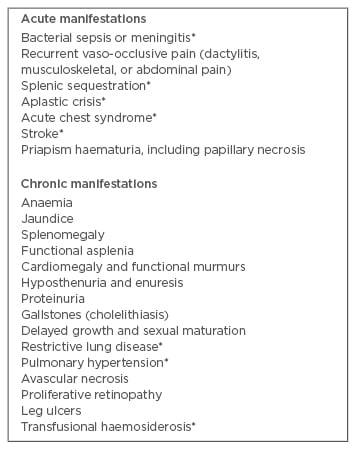
Table 2: Important clinical manifestations of sickle cell disease during childhood and adolescence.14
*Potential cause of mortality
MANAGEMENT OF SICKLE CELL DISEASE IN CHILDHOOD AND ACCESS TO MINIMUM STANDARDS OF CARE
In recent decades, advances in basic research and clinical investigation have produced a marked decrease in morbidity and mortality during early childhood.20-23 Comprehensive care including newborn screening, prophylactic penicillin, effective vaccinations (against pneumococcus, haemophilus, and annual influenza), stroke prevention programmes with transcranial Doppler (TCD) screening, chronic transfusion for children at risk of stroke, and disease-modifying treatments such as hydroxyurea (HU) have largely contributed to these results. This has virtually eliminated mortality from infections and stroke in British, French, and Belgian cohorts in the past decade.20-23 In contrast, mortality from sepsis and stroke is still an issue in areas where neonatal screening and stroke prevention programmes have been implemented more recently.24,25 The benefit of the previously described interventions (comprehensive care, newborn screening, antibiotic prophylaxis and vaccination, TCD screening, and HU) have been proven and they are considered as minimum standards of care for patients with SCD in several national European guidelines. In fact, the first action undertaken in many countries to address SCD has been the creation of a national network or group of experts for SCD and the development of national guidelines tailored to each country’s healthcare system characteristics and treatment availability.5,26-30 Such guidelines can be accessed online by paediatricians and paediatric haematologists, and address the management of acute complications (such as fever, vaso-occlusive painful crisis, acute chest syndrome, splenic sequestration) and chronic organ damage (pulmonary hypertension, kidney disease, cognitive impairment, etc.), detailing clinical pathways for accurate diagnosis and treatment.
The delivery of specialised, multidisciplinary, comprehensive care to a socially vulnerable population like the one constituted by patients with SCD in Europe, who are mainly first or second-generation immigrants, was challenging.31
Health operators in all countries were faced with language, cultural, and social barriers that in many cases were overcome by developing patient- centred services focussed on health education, with multilingual leaflets or booklets also available online (as in the case of the ones developed in France: www.rosfed.fr). A co-ordinated system of care between specialised centres, smaller hospitals, and primary care physicians is present in some areas and is useful to facilitate appropriate access to healthcare. Wherever implemented, comprehensive medical strategies, intended as global and holistic care, have reduced emergency and inpatient admissions while improving adherence to treatment both in the UK and Italy.31,32
NEWBORN SCREENING
Newborn screening allows early identification of affected patients, introduction of penicillin prophylaxis from 2 months of age, and reduction of mortality from infection, while allowing prompt enrolment in comprehensive care programmes. Universal newborn screening is available in the UK, Netherlands, several cities in Belgium, and Spain.5,7,33-36 Targeted screening for high-risk populations according to ethnic origin is available in France.37 Pilot newborn screening projects have been undertaken in Ireland, Italy, and Germany,8,38-43 but a national screening programmeis yet to be developed in these countries. Antenatal screening in the framework of anational programme for the prevention of haemoglobinopathies is present in Greece.2 Several obstacles or limits have been described in the implementation of the screening programmes;44-48 first and foremost the lack of funding from health authorities.8,40 Ethnic and racial issues due to the heritage of the second world war have also slowed the development of newborn screening or hampered the communication of test results to carriers in the Netherlands and Germany.29,44 Failure to identify affected patients in selective newborn screening due to misinterpretation of patient origin has also been described and raises the need for increased attention.44 After screening results are available, the management of patients and families varies across the countries, from being invited to genetic counselling45 to being notified and taken into paediatric SCD reference centres.4,5,8 Global comprehensive care after newborn screening is generally seen as the gold standard.
PENICILLIN PROPHYLAXIS AND VACCINATIONS
Penicillin prophylaxis and vaccination coverage have dramatically reduced the incidence of sepsis and mortality from infections in patients with SCD, not only in research studies but also in clinical care settings.20,31 The best results have been obtained in countries where newborn screening is available. While there is general agreement on the prescription of penicillin prophylaxis and vaccinations, few data are available in Europe on patient adherence to prophylaxis and vaccination coverage, as well as resistance to pneumococcus in children undergoing prophylaxis. Research in these areas could be useful to address possible barriers to treatment or to optimise prevention strategies. Annual influenza vaccination is also recommended in many guidelines, but the data on actual vaccination coverage from the various countries is scarce and does not always meet optimal standards.49-51
STROKE PREVENTION: TRANSCRANIAL DOPPLER AND CHRONIC TRANSFUSION
Extensive research has been conducted in Europe on the management of cerebrovascular complications in children with SCD.17,21,22 TCD for evaluation of intracranial circulation is widely recommended and recently, French and UK groups have included the screening of the extracranial circulation52-53 as part of the protocol for stroke prevention and reduction of silent cerebral infarcts. Despite the strong recommendation to perform TCD screening in every child with SCD starting at 2 years of age, <45% of children with SCD receive adequate screening with TCD in the UK.54 Data from other European countries are lacking. Several barriers to TCD screening have been identified, including: technical issues due to instrument availability and protocol standardisation (imaging versus non-imaging TCD), lack of trained personnel, and unfavourable schedules with TCD appointments on different days from haematology visits.31,55 Efforts should be made to investigate actual coverage of TCD screening and efficacy of stroke prevention in the remaining European countries in order to optimise treatment. A multicentre study focussing on the provision of TCD skills was delivered through an educational programme conducted in the UK, Ireland, and Italy, and demonstrated the success of a modular TCD training programme in achieving consistent, standardised, and comparable evaluation in three European countries.56
Patients at risk of stroke according to TCD results are offered chronic transfusion through simple top-up or exchange transfusions. The latter reduce iron overload, but are not available in every centre. While TCD allows identification of patients at risk of stroke and the ability to initiate appropriate treatment, it is not useful in screening for the other cerebrovascular complications of SCD, such as silent cerebral infarcts.
DISEASE-MODIFYING TREATMENTS: HYDROXYUREA, RED BLOOD CELL TRANSFUSION, AND BONE MARROW TRANSPLANTATION
HU is currently the only approved and routinely used drug for SCD. It reduces the frequency of acute vaso-occlusive painful crisis and acute chest syndrome, it improves organ function,5,28-29 and it has recently been demonstrated to have a role in stroke prevention.57 European centres have participated in proving the safety and benefits of HU in children with SCD.23,58-60 Currently, a study is underway to determine the long-term effects of HU in a European cohort.61 However, data regarding prescription of HU and adherence to HU treatment in Europe are lacking, and many publications coming from the USA report that there is an under-prescription and underuse of HU;62 this highlights caregiver and patient barriers, and is a field that warrants investigation. Some countries do not even have a paediatric formulation available, and have to acquire it from abroad or use inappropriate schedules, which is also an urgent issue to resolve.63
Red blood cell transfusions are a pivotal treatment of SCD, both for acute emergencies and chronic complications.5,27-29 Lack of ethnically matched donors is a concerning issue and extensive cell phenotype matching is recommended to avoid alloimmunisation.
The only curative option for SCD is bone marrow transplantation (BMT). The description of the indications for BMT, and of the different types of donor and conditioning regimens that can be used for BMT, goes beyond the scope of this review. It is worth noting, however, that several European centres have the resources to increase the possibility of performing BMT by using one of the parents as a donor (haploidentical transplantation).
OPEN ISSUES AND FUTURE PERSPECTIVES
The past few decades have seen service organisation for patients with SCD in several European countries. While high-level research is conducted in many centres and excellent care is routinely delivered, very few data report outcomes of healthcare delivery and utilisation across Europe. No up-to-date data on the burden of SCD across the EU is available, due to the absence of an organised system of data collection and to the lack of widespread newborn screening in many countries. Information on the rate of hospitalisation, length of stay, readmission rate, outpatient service utilisation, and outcome data, including mortality, are limited to some centres. A great deal of literature on healthcare utilisation, service delivery, and costs of care for SCD is available for the USA, but similar data have not yet been produced from Europe and this is a gap that should be filled to allow better service implementation.
Further aspects of care require co-ordinated action. Cerebrovascular complications are not limited to stroke prevention; silent infarcts and cognitive impairment pose a great burden on the health and quality of life of children with SCD, and will greatly impact them in later life.64 Investigations and interventions in this field have been performed in some countries but need to be expanded. Transition to adulthood is challenging65 and the mortality peak is shifting from early childhood to late adolescence and young adulthood. Adequate transition services and adult care are inconsistent across Europe.
In conclusion, as suggested by the EHA roadmap,16 optimising patient care and clinical trials to address basic aspects of clinical care are key issues in the field of SCD in Europe, together with new profiling of disease severity allowing personalised medicine and investigation of new therapeutic molecules for clinical management. These aspects will affect patient health outcomes and quality of life, as well as national and European healthcare systems by reducing hospital admissions, hospitalisation lengths, and care costs. A few practical suggestions, which are certainly not exhaustive, regarding issues to address and improve in the field of paediatric SCD are shown in Table 3.

Table 3: Practical suggestions for future perspectives.





