
Abstract
The myelodysplastic syndromes (MDS) form a heterogeneous group of clonal disorders with an increasing incidence in the elderly population and an emerging impact on healthcare resources. MDS are caused by gene mutations affecting the haematopoietic stem cells, leading to ineffective haematopoiesis, characterised by dysplasia and cytopenia, and a propensity to evolve towards secondary acute myeloid leukaemia (AML). Accurate diagnosis and risk assessment are essential for the correct treatment allocation. In lower-risk MDS patients, median survival reaches 3–8 years and mortality is mainly caused by cytopenia (cardiovascular events, infections, and bleeding). Therefore, the treatment for these patients should be focussed on reduction of disease-related complications, disease progression, and improvement of quality of life. In contrast, in higher-risk MDS patients, median survival ranges from 1–3 years and death from transformation to AML exceeds non-leukaemic mortality. Treatment should be aimed to delay progression to AML and improve overall survival. Allogeneic haematopoietic stem cell transplant remains the only curative option for higher-risk MDS patients. However, only a minority of patients are eligible for such intensive treatment. Consequently, most patients are managed with supportive care and palliative treatment, including growth factors, immune-modulators, and hypomethylating agents. Since elderly patients with chronic cytopenia are frequently seen in general practice, awareness of the wide spectrum of presentations of MDS and potential courses of lower and higher-risk diseases are important for primary healthcare physicians.
EPIDEMIOLOGY
Myelodysplastic syndrome (MDS) is predominantly diagnosed in elderly individuals at a median age >70 years. In European countries, the age-standardised incidence rate amounts to 2–3 per 100,000 patient years, with two-fold higher incidence rates in males compared to females. The only exception is found in MDS with deletion 5q, which has a female predominance.1,2-4 The age-specific incidence rate in patients >75 years rises over 10-fold, consistent with the general increase of cancer-incidence observed in the elderly. The age-adjusted incidence rate remained stable over recent decades, indicating that the observed increase of annual case-frequencies is mainly related to ageing and not to augmentation of age-specific risks.1 Due to an ageing population and increasing diagnostic awareness, MDS is likely to become an emerging haematological malignancy with a relevant impact on healthcare resources in the future.1
PATHOPHYSIOLOGY
MDS is a heterogeneous disease caused by accumulation of genetic lesions in the haematopoietic stem cell (HSC) compartment.5 The interaction of extrinsic genotoxic factors with intrinsic deficiencies in repairing DNA lesions, combined with loss of immunological tumour surveillance, are supposed to be responsible for the emergence and clonal progression towards acute myeloid leukaemia (AML).6 Genetic lesions initially identified in MDS were limited to structural chromosomal aberrations detectable by conventional cytogenetics. However, the understanding of genetic alterations associated with MDS/AML has rapidly evolved in recent years.7 Next-generation sequencing has allowed for the discovery of recurrent somatic driver mutations (RSDM), comprising genes of RNA-splicing factors, epigenetic regulators, transcription factors, cell-cycle regulators, cohesin complex factors, and cell-signalling molecules (Table 1).8-12 The molecular mechanisms and signalling pathways involved in the pathogenesis of MDS are very diverse and not fully understood. In lower-risk disease, an increased cell death rate is the hallmark of ineffective haematopoiesis and frequently involves the TP53 pathway.13 Interactions with cellular (e.g. T cells, natural killer cells, myeloid derived suppressor cells) and soluble factors (e.g. cytokines, danger signals), of the adaptive and innate immunity within the microenvironment are additional factors contributing to disease initiation and progression.14,15
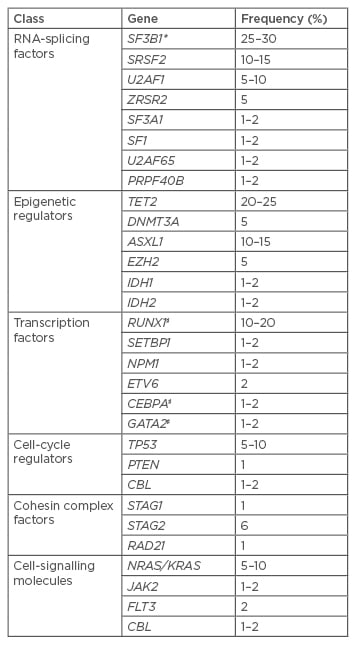
Table 1: Recurrently mutated driver genes in MDS.8-12
Selection of the most frequent genes affected by RSDM in MDS. Frequencies are only indicative, since they are extracted from studies which also included myeloid neoplasms other than MDS.
*High association with ringed sideroblasts; ♯Somatic and constitutional gene mutations.
RSDM: recurrent somatic driver mutations; MDS:myelodysplastic syndromes.
DIAGNOSTIC APPROACH
The diagnostic approach for primary MDS in adults has been reviewed in the European LeukemiaNet (ELN) recommendations.8 A thorough patient (exposures to genotoxic agents, pesticides, insecticides) and family history (signs for germ-line predisposition for patients <50 years of age) is complemented by a physical examination. Laboratory analyses are classified as ‘mandatory’ (peripheral blood, bone marrow aspirate/ biopsy, cytogenetic analysis), ‘recommended’ (fluorescence in situ hybridisation, fluorescence activated cell sorting), and ‘suggested’ for specific circumstances (single-nucleotide-polymorphism-array, mutational analysis).
Classification
MDS was initially classified according to the French–American–British (FAB) criteria. In 1997, a new classification system was proposed by the World Health Organization (WHO) and revised most recently in 2016.16 In this last revision, the historically used expression of ‘refractory anaemia’ was replaced by ‘MDS’, due to the fact that MDS patients may present with cytopenias other than anaemia. Classification of MDS considers the number of lineages affected by cytopenia and dysplasia, the presence of ring sideroblasts or the presence of mutations in SF3B1 (which is associated with ring sideroblasts), the number of blasts in the peripheral blood or bone marrow, and the presence of specific or MDS-defining cytogenetic alterations (e.g. del 5q) (Table 2).
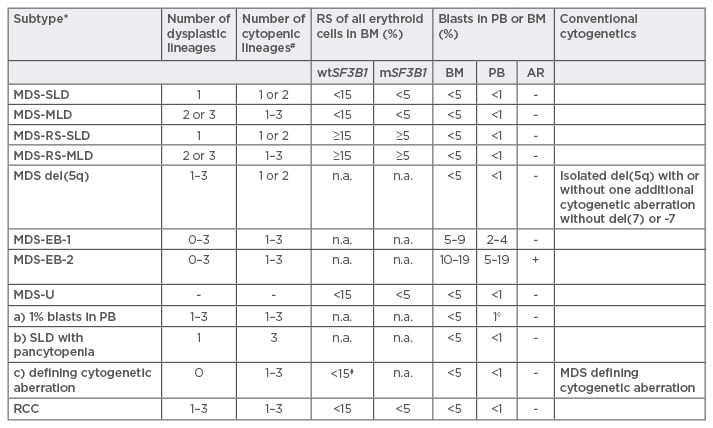
Table 2: World Health Organization (WHO) 2016 classification for myelodysplastic syndromes.16
*Without previous cytotoxic treatment or germline predisposition for myeloid neoplasms; #Cytopenias: haemoglobins <100 g/L, thrombocytes <100 g/L, neutrophils <1.8 g/L, monocytes <1 g/L; ◊1% blasts in PB must be confirmed with a second value; ⧫≥15%, corresponds to MDS-RS-SLD.
Caveat: If ≥50% are erythroid precursors and ≥20% blast cells of non-erythroid-lineage but <20% of all cells, this corresponds now to MDS (MDS-SLD/MLD or EB) and not any more to AML M6 erythroid/myeloid.
MDS: myelodysplastic syndromes; SLD: single lineage dysplasia; MLD: multi lineage dysplasia; RS: ringsideroblasts; EB: excess of blasts; RCC: refractory cytopenia of the childhood; wt/mSF3B1: wild type or mutated SF3B1; PB: peripheral blood; BM: bone marrow; AR: Auer rods.
Clonal Haematopoiesis in the Elderly
Unexplained anaemia is present in ~10–15% of patients >65 years of age. It is currently an active field of investigation to identify patients with ‘cryptic’ clonal haematopoiesis that may have an increased risk for progression towards overt haematological malignancies. Patients with cytopenia, but without sufficient dysplastic changes or MDS-defining cytogenetic alterations, are assigned to idiopathic cytopenia of unknown significance.17 In such circumstances, observation for 3–6 months with repetition of bone-marrow analysis prior to diagnosis of MDS is recommended.8 RSDM can be identified with an increased frequency in the elderly (10–20%). These individuals have normal peripheral blood values or only mild cytopenia that do otherwise not fulfill the diagnostic criteria for MDS. The condition is termed clonal haematopoiesis with indeterminate potential (CHIP: normal peripheral blood values with RSDM)18,19 or clonal cytopenia with unknown significance (CCUS: cytopenia with RSDM),20 respectively. The possibility to use next-generation sequencing to identify clonal signatures allows the identification of patients at early stages and at risk for development of overt haematological malignancies.
GENERAL ASPECTS OF MYELODYSPLASTIC SYNDROME PATIENT MANAGEMENT
MDSs are heterogeneous diseases and the natural course varies from a chronic, indolent state with asymptomatic cytopenia, to a condition with rapid evolution towards secondary AML. Two-thirds of patients with MDS die of complications related to cytopenia and one-third succumb to AML progression.21 Management is based on disease and patient-related factors using scoring systems that have been validated in MDS patients. Besides age, other patient-related factors include comorbidities and frailty, which are increasingly recognised as important risk factors, especially in elderly patients.8 Accurate diagnosis and risk-assessment are, therefore, essential for appropriate treatment allocation. In lower-risk MDS patients, median survival reaches 3–8 years and mortality is mainly caused by cytopenia, (cardiovascular events, infections, and bleeding.) Therefore, the treatment for these patients should be focussed on reduction of disease-related complications, disease progression, and improvement of quality of life.22,23 In contrast, in higher-risk MDS patients, median survival ranges from 1–3 years and transformation to AML exceeds non-leukaemic mortality. Treatment should be aimed to delay progression to AML and improve overall survival.24,25
Disease-Related Risk Factors
The risk for progression to AML and overall survival can be determined by the International Prognostic Scoring System (IPSS), the revised IPSS (IPSS-R), and the WHO Prognostic Scoring System (WPSS).26-28 The numbers of blood lineages affected by cytopenia are used in all prognostic-scoring systems. Blast count and type of cytogenetic alterations are relevant for the IPSS and the IPSS-R, whereas the WPSS uses the WHO MDS subtype and information on transfusion dependency. Less advanced MDS is characterised by <5% of blasts in the bone marrow, no circulating blasts in the peripheral blood, isolated anaemia, transfusion independency, and normal karyotype or favourable cytogenetic alterations. In contrast, advanced MDS is defined by >5% blasts in the bone marrow, circulating blasts, cytopenia of multiple lineages, poor-risk or complex cytogenetic alterations, and transfusion dependency.
Patient-Related Risk Factors
In oncological treatment, it is essential to balance efficacy against tolerability, which is particularly important for elderly and frail patients. Karnofsky and Eastern Cooperative Oncology Group (ECOG) scores can be used to assess the performance status. However, these are influenced by age and, therefore, are insufficient to assess comorbidity and frailty. The Charlson comorbidity index (CCI) was adapted by Sorror et al. for patients undergoing allo-haematopoietic stem cell transplantation (HSCT)29,30 and also validated for MDS-patients (HCT-CI).31 The Italian MDS Study Group simplified this scoring system for MDS patients (MDS-CI) by considering only cardiac, hepatic, pulmonary, and renal comorbidities as well as prior treatment for solid tumours.32 Frailty and functionality in daily living can be scored with general geriatric assessment tools. Based on an increasing cancer frequency in elderly patients, as well as a rising number of targeted therapies with specific side effects, assessment of patient-related factors is an emerging requirement for appropriate treatment allocation.
Treatment Approach
Due to the complexity of the disease management, MDS patients with symptomatic cytopenia should be assessed and followed by physicians with experience in the management of MDS. Moreover, these patients should be included into prospective registries and offered clinical trials, if they fail to respond to standard treatment.
Lower-Risk Myelodysplastic Syndrome Patients (Figure 1A)
Watchful-waiting
Life expectancy of patients >70 years of age with MDS-SLD or MDS with del(5q) is not significantly shorter than an age-matched elderly population.28 Patients with low/intermediate-1 IPSS and asymptomatic cytopenia should, therefore, be followed regularly without any treatment.8 This recommendation might change in the future, as molecular testing will recognise higher-risk diseases and novel drugs will enter into the therapeutic armamentarium that may delay progression.
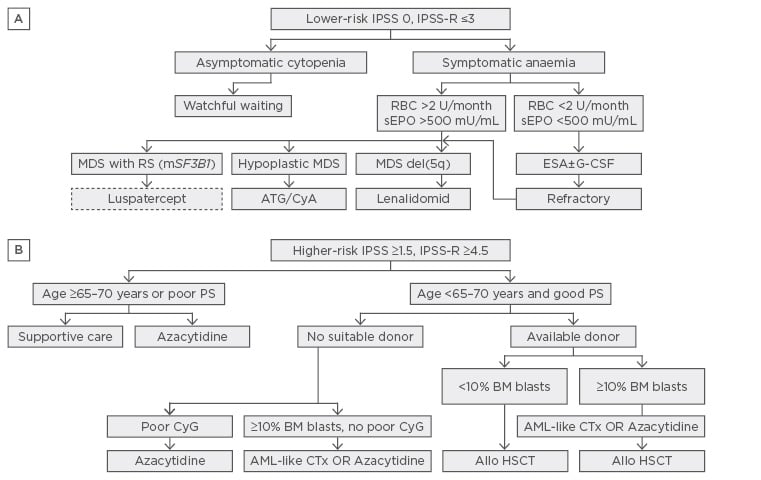
Figure 1: Treatment algorithm for lower and higher-risk myelodysplastic syndromes.8
Allo-HSCT: allogeneic haematopoietic stem cell transplantation; ATG: anti-thymocytic globulin; BM: bone marrow; CSA: cyclosporin A; CTx: chemotherapy; ESA: erythropoietin stimulating agent; G-CSF: granulocytic colony stimulating factor; mSF3B1: mutated SF3B1; PB: peripheral blood; PS: performance status; RBC: red blood cell concentrate; RS: Ringed sideroblasts; sEpo: serum erythropoietin.
Supportive treatment
Supportive treatment consists of transfusions, infection prophylaxis, antiemetics, analgesics, iron chelation, and growth factors. Transfusion dependency is generally associated with advanced disease stage and worse outcome.28 Many studies have been published trying to define the most appropriate triggers for transfusions. Transfusions of red blood cells (RBC) have been shown to improve quality of life and thrombocyte concentrates have reduced bleeding events. The current general agreement is to transfuse RBC if haemoglobin-levels drop <80 g/L, or at higher levels if patients are symptomatic due to age or other comorbidities. Thrombocyte concentrates are prophylactically transfused if thrombocytes drop <5–10 g/L, or <20 g/L if additional risk factors, such as fever or mucositis, are present. Thrombocytes <50 g/L are associated with a higher bleeding risk in patients treated with anticoagulants, although MDS patients may harbour thrombocyte dysfunctions or other haemorrhagic diathesis and present with major bleeding even at higher values. Transfusions are generally associated with a higher risk of adverse events such as alloimmunisation and transfusion-associated complications.33 RBC are generally irradiated in patients that are potential candidates for allo-HSCT, in order to reduce risk of HLA alloimmunisation and transfusion-associated graft versus host disease. Controlled trials supporting this approach are, however, lacking.
In heavily transfused patients with >15–20 RBC and a ferritin level >1,000 mg/L, iron chelation can prevent iron-overload, improve cytopenia, and may have an impact on survival.34-36 However, it remains a matter of debate which patients should be chelated and at what stage of the disease, since results from randomised studies are still lacking in MDS patients.37 Chelation is generally offered with deferasirox (Exjade®) to patients that are candidates for allo-HSCT or with a life expectancy >1 year.
Patients with low/intermediate-1 IPSS, with haemoglobin-levels <100 g/L, serum erythropoietin <200–500 U/L, red cell transfusion independency, or <2 RBC units/month are candidates for treatment with erythropoietin stimulating agents (ESAs).34,38,39 No relevant differences have been identified between distinct ESA products (recombinant human erythropoietin alpha and beta or darbepoietin alpha). The required erythropoietin dose levels are higher compared to patients with renal insufficiency and are usually started at 30,000 U/week subcutaneously (~150 ug darbepoietin alpha). The dose is doubled after 6–8 weeks in cases of lack of response. Current guidelines suggest addition of granulocytic-colony stimulating factor (G-CSF) (at 1-3×300–480 ug/week subcutaneously) in those patients with low/ intermediate-1 IPSS unresponsive to ESA within 8–12 weeks.34,40,41 However, large, randomised studies supporting this recommendation, especially compared to high-dose ESA (60,000 U/week), are lacking. The adherence to a strict ESA/G-CSF substitution regime is important in order to rapidly identify refractory patients that harbor a worse prognosis and may be candidates for allo-HSCT or clinical trials. Thrombopoietin-stimulating agents (TSA) (i.e. romiplostim, eltrombopag) are currently tested in clinical trials in patients with thrombocytopenia and low/intermediate-1 IPSS. TSA show a beneficial effect on platelet count and reduction in bleeding without an impact on overall survival.42,43 It is important to note that ESA and G-CSF are approved for the treatment of MDS patients; however, the conditions for reimbursement are highly heterogeneous in distinct European countries.
Disease-modifying treatment
Immunomodulatory drugs
The anti-cytokine and anti-angiogenetic effect of thalidomide was identified to reduce transfusion need in MDS patients.44 Due to the long-term neurologic toxicity of thalidomide, a 4-amino-glutarimide derivate, lenalidomide (Revlimid®), lacking this side effect, was developed and showed sustained transfusion independency and cytogenetic responses in about half of all MDS patients with low/intermediate-1 IPSS and isolated del(5q).45 Response was dependent on additional cytogenetic anomalies and transfusion dependency. In a Phase III study, about a quarter of non-del(5q) MDS patients with low/intermediate-1 IPSS achieved also transfusion independency, whereas mutations in TP53 were associated with resistance and disease progression.46,47 The ELN guidelines recommend lenalidomide for treatment of i) low/ intermediate-1 IPSS MDS patients with isolated del(5q) after failure of ESA treatment, and ii) in del(5q) MDS patients with intermediate-1 IPSS due to additional cytogenetic anomalies or excess of blast. In del(5q) MDS patients with evidence of TP53 mutations the choice remains open between lenalidomide and other therapies, including hypomethylating agents (HMA) and allo-HSCT.34
Immunosuppressive treatment
Ten percent of MDS patients present with hypoplastic bone marrow and are potential candidates for immunosuppressive treatment with antithymocyte globulin (ATG) combined with cyclosporine A (CyA). Response rates of around one-third can be achieved and responders can be predicted based on absence of blasts or ring sideroblasts, young age (<60 years), low IPSS, short duration of transfusion requirement, and HLA-DR15 phenotype.48 The current ELN guidelines recommend ATG with 6 months of CyA in younger MDS, especially if hypoplastic bone marrow and favourable response features are present. No recommendation could be made on the duration of maintenance therapy in patients responding to ATG/CyA.34 The combination with the TSA eltrombopag has been shown to improve response rates in aplastic anaemia, but is not approved for hypoplastic MDS.49
Higher-Risk Myelodysplastic Syndrome Patients (Figure 1B)
Hypomethylating agents
MDS patients with excess of blasts or higher-risk scores that are ineligible for intensive chemotherapy and allo-HSCT are candidates for palliative treatment with HMA. The pyrimidine nucleoside analogs 5-azacytidine (AZA) and 5-aza-2’deoxycytidine/decitabine have been studied in Phase III studies in higher-risk MDS patients.50,51 HMA are generally well-tolerated and showed significant higher partial and complete responses compared to best supportive care including hydroxyurea and low-dose cytosine arabinoside (AraC). However, HMA remain inferior to more intensive induction chemotherapy followed by allo-HSCT, for which, however, only a minority of elderly patients are eligible. MDS with complex karyotype, either in elderly or younger patients without suitable stem-cell donor, should preferentially be treated with HMA, due to the lower rates of complete remission and higher toxicity imparted by intensive chemotherapy.52 There are no generally accepted predictive molecular markers for response to HMA and also the duration of treatment remains unclear. Lower response rates to AZA have been described for patients with previous treatment with AraC, higher numbers of bone marrow blasts (>15%) and abnormal or complex karyotype. Lower overall survival was found in patients with poor performance status (ECOG >2), higher cytogenetic risk scores, circulating blasts, and high transfusion requirement (>4 units over 8 weeks).53 No established treatment is currently available after failure to HMA and these patients should be offered clinical trials.
Intensive chemotherapy
Standard AML-based chemotherapy is reserved for high-risk MDS patients that are eligible for induction chemotherapy before proceeding to allo-HSCT. Younger age, good performance status, and favourable cytogenetics are independent prognostic factors associated with improved survival.54 The ELN guidelines recommend induction chemotherapy for fit patients, <65–70 years of age, without a suitable donor and >10% bone marrow blasts but without adverse cytogenetic characteristics.8 Patients with adverse or complex cytogenetic, as well as mutations or deletions in TP53, have a worse response to intensive chemotherapy and may be offered treatment with HMA with or without subsequent allo-HSCT.52,55 The current evidence is insufficient to recommend HMA for induction prior allo-HSCT outside of clinical trials.8
Allogeneic haematopoietic stem cell transplantation
Allo-HSCT remains the only curative option but is only appropriate for MDS patients considered fit for intensive therapy. Assessment of comorbidities is important for deciding which patients should be referred for allo-HSCT. The HCT-CI can estimate non-relapse mortality and is used for this purpose.30 Cumulative incidence of treatment-related mortality (TRM) after allo-HSCT with HLA-identical siblings ranges from 32% at 1 year to 37% at 3 years, the cumulative incidence of relapse from 17% at 1 year to 23% at 3 years, and disease-free survival from 53% at 1 year to 40% at 3 years. The international bone marrow transplant registry (IBMTR) examined different strategies for patients with newly diagnosed MDS. They found that delayed transplantation was associated with maximal life expectancy, whereas for intermediate-2/high IPSS, immediate transplantation was best.56 Age is the most important predictive factor for overall survival. A retrospective analysis of the European Group for Blood and Marrow Transplantation (EBMT) found a TRM of 30% in patients <20 years of age, 43% in patients 20–40 years of age, and 50% in patients >50 years of age.57 However, recent changes in conditioning and careful patient selection have shown that allo-HSCT is also feasible in patients between 60 and 70 years of age. The ELN guidelines recommend allo-HSCT for fit patients ≤65–70 years of age with intermediate-2/high-risk IPSS and those with intermediate-1 IPSS with excess of blasts or poor risk cytogenetics. For MDS patients not eligible for myeloablative conditioning due to comorbidities, reduced intensity conditioning allo-HSCT should be considered, preferably within clinical trials. The EBMT is currently recruiting AML/MDS patients for a prospective, randomised study to evaluate the significance of allo-HSCT with reduced intensity conditioning in patients between 60 and 75 years of age.
FUTURE DIRECTIONS
The discovery of RSDM opens a broad range of novel opportunities by tracking longitudinally clonal evolution at early stages of clonal haematopoiesis (CHIP and CCUS). Changing classifications and the evolving diagnostic tools make disease-based risk assessment in MDS patients a dynamic and evolving field. Mutations in TP53, EZH2, ETV6, RUNX1, and ASXL1 have been shown to confer worse prognosis and the presence of mutations in these genes increased the accuracy of risk assessment, especially in lower/intermediate-1 IPSS patients. A molecular based scoring system is currently under development and may be incorporated into a novel, revised IPSS (IPSS-R mole).58 Lower-risk MDS patients with ring sideroblasts or SF3B1 mutations (MDS-RS-SLD/MLS) have been shown to respond to luspatercept. This drug scavenges tumour growth factor (TGF)-β superfamily ligands and improves erythropoiesis by mechanisms distinct from erythropoietin. In a Phase II study, luspatercept showed interesting erythroid responses ranging from 50–90% and led to transfusion independency in 30–60% of ESA-refractory patients.59 Based on the assumption that low-dose and longer exposure of HMA might be more efficient in improving differentiation, oral AZA formulations for a prolonged period of time are currently being tested in clinical studies in lower-risk IPSS MDS patients.60 Other drugs that are investigated include toll-like receptor 2 antibodies, CD95 ligand (FAS-ligand) inhibitors, multikinase inhibitors (e.g. rigosertib), checkpoint inhibitors (e.g. nivolumab, durvalumab), telomerase inhibitors (e.g. imetelstat), and inhibitors of mutated IDH1/2 (e.g. AG-120/AG-221). Moreover, improvement of treatment allocation based on efficacy, tolerability, and affordability remains an active field of investigation in the context of health services research. The inclusion of MDS patients in prospective national and international registries is, therefore, highly encouraged. Longitudinal observational studies of patients at early stages of clonal haematopoiesis with integration of detailed health-related and molecular data will be important not only to improve our understanding of disease biology, but also for prognostication, prediction, and allocation to treatment.





