
Abstract
Multiple myeloma (MM) continues to be an incurable disease impacting mainly an ageing population. Comorbidities, disease characteristics, and drug toxicity profiles heavily influence treatment selections. Despite single agent activity of many anti-MM agents, opportunities to maintain responses most often include combination therapy with immunomodulator and/or proteasome inhibitor therapies. Monoclonal antibodies (moAb) have become an additional backbone to both newly diagnosed and relapsed or refractory transplant eligible and ineligible patients. Tolerability of these agents offers an additional benefit particularly to an ageing population. Two newly approved moAb targeting CD38 and B-cell maturation antigen have been added to the anti-MM arsenal. Isatuximab, a chimeric anti-CD38 moAb, is the second U.S. Food and Drug Administration (FDA)-approved CD38 targeted therapy offering unique mechanisms of action owing to differences in epitope binding and favourable side effect profiles. Belantamab mafodotin, a B-cell maturation antigen drug-antibody conjugate, is a first-in-class humanised moAb containing a distinct microtubule-disrupting agent: monomethyl auristatin-F. Its distinctive anti-MM activity includes antibody-dependent cellular cytotoxicity and phagocytosis, as well as direct cytotoxicity caused by internalisation of monomethyl auristatin-F. This review focusses primarily on the mechanisms of action, resistance patterns, and clinical utility of two recently FDA-approved agents; isatuximab in combination with pomalidomide and dexamethasone for relapsed or refractory MM exposed to at least two or more lines of therapy, and belantamab mafodotin monotherapy in relapsed or refractory MM exposed to four or more lines of therapy.
INTRODUCTION
As the second most common haematological malignancy, multiple myeloma (MM), a plasma cell disorder, continues to affect a significant portion of patients with increasing incidence over the past 25 years.1–3 MM remains an incurable disease despite the continued improvement of outcomes over the past decades with immunomodulatory (IMID) and proteosome inhibitor (PI) therapies. Targeted immunotherapy with monoclonal antibodies (moAb) is critical for the successful treatment of different malignancies and is no different in MM. The first-in-class, humanised IgG1-κ moAb daratumumab targets the CD38 epitope and has rapidly changed the treatment landscape of MM, moving quickly from the relapsed or refractory to the upfront setting with unparalleled results.4-6
Several anti-CD38 and B-cell maturation antigen (BCMA) therapies are being developed for the treatment of MM.7 Reflecting this rapid progress of MM drug development, two new agents received approval from the U.S. Food and Drug Administration (FDA): belantamab mafodotin, a monotherapy BCMA-directed antibody and microtubule inhibitor conjugate, and isatuximab, a CD38-directed cytolytic antibody, in combination with pomalidomide and dexamethasone. Studies have shown that these therapies are effective in the treatment of relapsed or refractory MM (RRMM) and are well tolerated with manageable side effects.8,9
This review specifically focusses on isatuximab and belantamab mafodotin including discussion of the mechanisms of action, clinical trial development leading to FDA approval, clinical activity, relevant adverse events (AE), as well as the manner in which these two agents are used within the MM treatment landscape. There is also a brief discussion of emerging moAb for the management of MM.
CD38 MONOCLONAL ANTIBODIES: ONE TARGET, MANY ACTIONS
Immunotherapy for the treatment of MM has become a significant addition to the anti-myeloma repertoire. CD38, a Type II transmembrane glycoprotein with ectoenzyme properties, has a prominent role in proliferation and growth of MM cells.10,11 CD38 directed moAb offer a unique target because of its higher presence on MM and plasma cells, while having low expression on other myeloid and lymphoid cells, making it an ideal anti-MM therapy candidate.2 Several mechanisms of anti-CD38 activity have been shown and are dependent on fragment crystallisable (Fc)-based immune activation. These include complement-dependent cytotoxicity (CDC), antibody-dependent cellular cytotoxicity (ADCC), and antibody-dependent cellular phagocytosis (ADCP), as well as direct apoptosis. CD38 moAb additionally offer immunomodulatory effects by way of regulatory and suppressor cell reduction and improved antitumour activity(Figure 1A).12-14 Differences among anti-CD38 moAb are highly dependent on the Fc-directed activity driven by location of epitope binding and subsequent anti-MM potency of the many immune mechanisms. There are currently four CD38 moAb therapies with Phase II or more clinical data (daratumumab, isatuximab, MOR,15 and TAK-07916).10,17

Figure 1: A) Anti-CD38 tumour kill mechanisms; B) belantamab mafodotin anti-multiple myeloma targets.
ADCC: antibody-dependent cellular cytotoxicity; ADCP: antibody-dependent cellular phagocytosis; BCMA: B-cell maturation antigen; CDC: complement-dependent cytotoxicity; MMAF: monomethyl auristatin-F; moAB: monoclonal antibody; NK: natural killer.
Mechanisms and Resistance Methods
Effector cells, particularly natural killer (NK) cells are a crucial player in ADCC, whereas monocytes and macrophages are integral in ADCP-mediated cell killing and are the drivers at the forefront of anti-CD 38 moAb activity. Complement-driven activation increases recruitment of other immune cells and phagocytosis, while reducing inhibitory immune effector cells. Unlike other anti-CD38 moAb, daratumumab exhibits stronger CDC-driven activity over all other anti-MM mechanisms.13 Direct anti-MM properties are highly variable among CD38 moAb and are dependent upon the ability to trigger apoptosis without Fc-driven binding. Daratumumab, unlike isatuximab, requires secondary cross-linking to induce programmed cell death, whereas isatuximab independently induces reactive oxygen species and liposomal-mediated death.10,13 Suppression of regulatory cells, including T and B cells, leads to improved effector (T and NK) cell numbers and activity while promoting an anti-MM microenvironment.12,13
Eventually patients with MM exposed to CD38 directed therapy will progress; however, several resistance mechanisms, both primary and acquired, are known and strategies to overcome this barrier are expanding.12 Concerns over prior therapy exposure, disease cytogenetics, and reintroduction of prior refractory agents are some patient-specific factors to consider.12 In a study conducted by Nijhof et al.,18 evaluation of Fc-dependent mechanisms of daratumumab showed heterogeneous CD38 expression, but similar activity of ADCC and CDC among newly diagnosed MM (NDMM) and RRMM patients, indicated that refractoriness to other anti-MM agents did not confer similar refractory response with CD38 moAb.18 Utilisation of anti-CD38 therapy in smouldering MM (SMM) showed higher single-agent daratumumab response-rates compared to heavily pretreated MM patients, leading the authors to note potential variations in the tumour microenvironment and immune impairment with disease progression.19 Single-agent activity of anti-CD38 therapies is evident; however, durability of response is limited. Combination of anti-MM therapy (PI or IMID) is one possible mechanism to boost activity and potentially overcome resistance. Synergistic activity of IMID therapy to augment immune effector cell activity and enhance anti-CD38 therapy has been shown in patients who were previously refractory to one or both therapies.20-23 Additionally, high-risk cytogenetic features such as t(4;14), t(4;16), and del17p continue to negatively impact overall response and survival of patients with MM. Anti-CD38 moAb therapy unfortunately does not abrogate inferior responses in high-risk patients.12,21,24
Additional resistance to CD38 moAb therapy may develop as a result of loss of CD38 cell surface density expression and/or increase in soluble CD38, leading to reduced ADCC, ADCP, and CDC capabilities.10,13 Susceptibility to these killing methods is heavily driven by cell surface expression; methods to increase surface density with all-trans retinoic acid,18 panobinostat,25 and IMID therapies have been proven to upregulate CD38 expression.12 Upon administration of daratumumab, a significant reduction in CD38 density expression from direct cell surface loss has been previously noted, regardless of treatment response.11,13 This reduction can be transient as baseline levels have been restored up to 6 months postexposure.18 This phenomenon is not noted with isatuximab therapy, although an increase in CD38 internalisation has been reported, this property is potentially driven by a different epitope binding site.10 Other drivers of resistance may include complement inhibitory proteins, anti-apoptotic proteins such as survivin, reduction in NK-cell numbers, and Fc-receptor polymorphisms.13
ISATUXIMAB: EXPANSION OF CD38 TARGETING
Unlike other CD38 moAb therapies, isatuximab’s epitope binding on CD38 is located away from catalytic activity site, uniquely increasing its inhibition of enzymatic activity and contributing to its variances in mechanism of action.10 Isatuximab’s main anti-MM kill mechanism is through ADCC, which can be enhanced through concurrent use of both PI and IMID therapies. Demonstrations of enhancement in direct cytotoxicity and cell lysis have been shown with combined therapies versus single-agent isatuximab; although, isatuximab does exhibit a dose-dependent CD38 enzymatic inhibition more potent than daratumumab.10,26 During initial monotherapy dose-escalation trials, isatuximab was given intravenously to RRMM patients at dosages up to 20 mg/kg weekly or every 2 weeks. The maximum tolerated dose was not reached because of lack of significant AE. The most common AE was infusion-related reactions (IRR) occurring in 49.3% patients, with mandatory prophylaxis given at dose levels more than 3 mg/kg. Symptoms included shortness of breath, nausea, headache, chest discomfort, and pyrexia. Most often, IRR occurred within first infusion and was associated with CDC activity. Haematologic toxicity occurred in 45–98% of patients, while fatigue and nausea occurred in approximately 35%.27 A Phase II dose-escalation trial28 supported isatuximab clinical and pharmacokinetic activity and determined the single-agent dosing strategy of 20 mg/kg weekly for four doses, followed by 20 mg/kg every 2 weeks. When combined with dexamethasone 40 mg or 20 mg daily in patients aged >75 years, isatuximab combination therapy had significant improvements in overall response rate (ORR), median progression-free survival (PFS), and reduction in IRR.29
Synergistic activity with other anti-MM therapies has proven effective throughout the history of MM treatment. Due to several MM subclones and the heterogeneity of the disease, combination strategies with IMID and PI have been effective with daratumumab and now isatuximab.26 Isatuximab combination with other IMID, such as lenalidomide, established a lower dosing strategy of four weekly doses of 10 mg/kg followed by doses every 2 weeks because of similar outcomes but higher Grade ≥3 toxicities noted with higher dosing.21 Most recently, isatuximab was studied in a Phase II trial24 combining pomalidomide in patients with lenalidomide and PI refractory disease (82% and 84%, respectively). These results led to a Phase III, prospective trial (ICARIA-MM)8 of isatuximab with and without pomalidomide and dexamethasone in RRMM patients. Triple therapy in highly refractory patients improved PFS regardless of age (>75 years),30 renal impairment,31 high-risk cytogenetics,32 or those with more than three prior lines of therapy33 or dual-refractory disease. Minimal residual disease negativity (level of 10-5) was also obtained in 5% of triple therapy patients versus none in doublet combination patients. Neutropenia and infections remained the most common AE among both treated groups with approximately 40% (Grade 3–4: 2.6%) experiencing isatuximab-IRR.8 Outcomes from the ICARIA trial8 led to FDA approval of isatuximab34 on 2nd March 2020. Clinical discussion of the major isatuximab trials is summarised in Table 1. 8,21,24,27,28
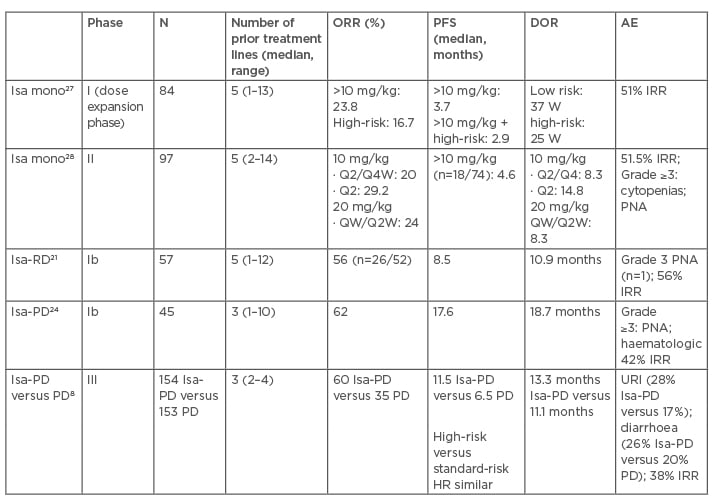
Table 1: Isatuximab clinical trials in relapsed or refractory multiple myeloma patients.8,21,24,27,28
AE: adverse event; D: dexamethasone; DOR: duration of response; HR: hazard ratio; IRR: infusion-related reactions; Isa: isatuximab; mono: monotherapy; ORR: overall response rate; PD: pomalidomide and dexamethasone; PFS: progression-free survival; PNA: pneumonia; Q: every; R: lenalidomide; URI: upper respiratory infection; W: weeks.
Additional activity was also noted with a PI in a Phase Ib trial of isatuximab with carfilzomib and dexamethasone. When given together with biweekly carfilzomib, isatuximab had an ORR of 60.6% in patients who had received prior carfilzomib (45.0%) and were dual-refractory (79.0%).35 Similar to the ICARIA clinical trial, the IKEMA study is prospectively evaluating isatuximab with and without carfilzomib and dexamethasone.36 Results of this trial will further add to the understanding of standard doublet-therapy versus triple-therapy in RRMM patients, especially amongst those with dual-refractory disease.
CLINICAL TRIALS AND FUTURE OPPORTUNITIES
Opportunities to improve patient care experiences with different CD38 moAb include recent approval of subcutaneous daratumumab administration,37 isatuximab fixed-volume infusions reducing infusion duration to less than 2 hours,38 reduced IRR moAb MOR-202,15 and nonhyaluronidase-containing subcutaneous formulation of TAK-079.16 Due to its efficacy and tolerability in the RRMM setting, there are several ongoing clinical trials investigating the efficacy of isatuximab in combination with both IMID and PI therapies in patients with NDMM. These include both transplant eligible39 and transplant ineligible40-43 patients. Additionally, isatuximab is currently being investigated as a monotherapy in SMM.10,26 Utilising CD38 moAb therapy earlier on may benefit from a more ‘fit’ immune system for modulation; however, exact sequence of use and corresponding resistance patterns are still being questioned.
Concerns regarding emergence of CD38 moAb therapies in both SMM and NDMM patients may change these responses in RRMM patients, however, full understanding has yet to be elucidated. Additionally, with different mechanisms of action and epitope binding properties, isatuximab may be useful in previously exposed daratumumab patients. Gandhi and colleagues23 reported on the extent of success of anti-CD38 therapy in 275 patients with refractory CD38 disease. This retrospective study (MAMMOTH) analysed triple-, quadruple-, and penta-refractory patients exposed to daratumumab (93%) and isatuximab (7%). Survival was significantly affected by level of refractoriness to anti-MM therapies. Patients who were penta-refractory had approximately 5.0 months less survival compared with nontriple-refractory (11.2 months) and those refractory to CD38 moAb had an overall survival (OS) of 8.6 months. A Phase I trial44 of isatuximab monotherapy in patients with prior daratumumab exposure was ongoing at the time of writing.
Clinical responses with anti-CD38 therapies are promising; however, due to small numbers of high-risk patients in clinical trials (20% approximately in monotherapy27 and ICARIA-MM8), it is not fully clear if anti-CD38 therapies can mitigate these oncogenic impacts on durability of response.27 A trial45 is currently investigating the role of isatuximab monotherapy in high-risk SMM patients and may offer further insight into the role of targeted therapies in modifications of patient-specific therapy based on mutations, molecular characteristics, and minimal residual disease status,14 and may also offer insight into the particular role of CD38 moAb therapy to obtain deeper and better responses.
B-cell Maturation Antigen: A New Versatile Target
Despite significant advances in MM treatment, many patients develop resistance or intolerance to available therapeutics including PI, IMID, and anti-CD38 moAbs. In the last few years, BCMA has emerged as an appealing target in MM because it is almost exclusively expressed on mature B-lymphocytes, plasma cells, and MM cells. BCMA belongs to the TNF receptor superfamily and is critical for bone marrow plasma cell long-term survival.46 Serum BCMA concentrations have been shown to be elevated in patients diagnosed with MM compared with healthy controls and patients with more progressive disease as opposed to those responding to treatment.47,48 These observations have led to various treatment modalities being investigated targeting BCMA, including antibody drug conjugates (ADC), chimeric antigen receptor T cells (CAR-T), and T-cell engaging bispecific antibodies.49 For the purpose of this review, advances in ADC therapies targeting BCMA will be outlined with a focus on the recently FDA-approved belantamab mafodotin.
Belantamab Mafodotin: One More Card to Play
Belantamab mafodotin has a multimodal activity against MM cell lines (Figure 1B). It is a humanised, afucosylated IgG1 anti-BCMA moAb connected by a protease-resistant linker to monomethyl auristatin-F (MMAF), a microtubule polymerisation inhibitor. Upon binding to BCMA, belantamab mafodotin is rapidly internalised, releasing its MMAF toxic payload and triggering cell cycle arrest in the G2/M phase that is followed by apoptosis.50 In addition, the afucosylation of Fc tail increases binding affinity of belantamab mafodotin to FcγRIIIa receptors present on target cells leading to ADCC and ADCP.51
Based on promising preclinical data, belantamab mafodotin was evaluated in a dose-escalation and expansion Phase I DREAMM-1 study in RRMM patients who received prior treatment with alkylators, PI, and IMID, and were resistant to the latest line of therapy.52,53 In the dose-expansion phase, ORR was 60%, including: two patients (6%) with stringent complete response; three patients (9%) with complete response; 14 patients (40%) with very good partial response (VGPR); and two patients (6%) with partial response. Median time to response was 1.2 months, with responses deepening with time. Median PFS was noted to be 12.0 months with a median duration of response (DOR) of 14.3 months. Of significance, median PFS of 6.2 months was much lower in patients who had received prior daratumumab therapy and were refractory to PI and IMID. Grade 3 or 4 AE were observed in 83% of patients including thrombocytopenia (35%) and anaemia (17%). Corneal toxicity, both keratopathy and visual acuity changes, occurred in 69% of patients and was likely related to the direct effects of MMAF-related toxicity. Twenty-nine percent of patients experienced IRR, the majority of them being mild in severity and occurred with first dose. Recently, Lonial and colleagues9 published the results of the DREAMM-2 study, a two-arm open-label randomised Phase II trial that evaluated belantamab mafodotin 2.5 mg/kg versus 3.4 mg/kg given intravenously every 3 weeks in RRMM patients who had received at least three prior lines of therapy and were refractory to PI and IMID, and refractory or intolerant to anti-CD38 moAb. ORR was 31% in the 2.5 mg/kg arm versus 34% in the 3.4 mg/kg arm with at least VGPR observed in 19% and 20%, respectively. The median PFS reported was 2.9 months in the 2.5 mg/kg group versus 4.9 months in the 3.4 mg/kg arm. The median DOR and OS data are not mature to date. The most common Grade 3 or 4 AE were keratopathy (27% in 2.5 mg/kg versus 21% in 3.4 mg/kg), thrombocytopenia (20% versus 33%, respectively), and anaemia (20% versus 25%, respectively). The findings of the dose-expansion part of DREAMM-1 and DREAMM-2 trials are summarised in Table 2.9,52,53
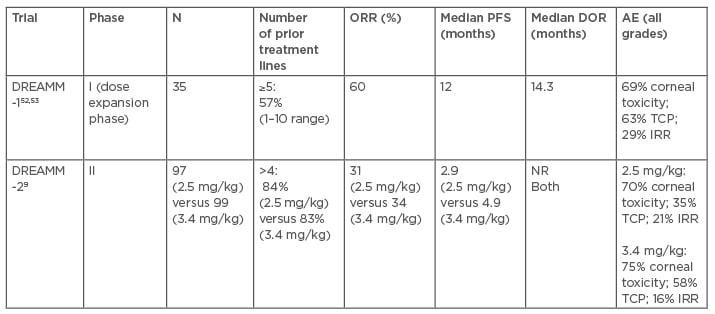
Table 2: Belantamab mafodotin clinical trials with relapsed or refractory multiple myeloma patients.9,52,53
AE: adverse event; DOR: duration of response; IRR: infusion-related; NR: not reached; ORR: overall response rate; reaction; PFS: progression-free survival; TCP: thrombocytopenia.
DREAMM-2 trial outcomes in patients with high-risk cytogenetics including t(4;14), t(14;16), 17p13del, or 1q21+ have been reported separately.54 High-risk cytogenetics were observed in 42% of patients in the 2.5 mg/kg group and 47% of patients in the 3.4 mg/kg group. ORR was reported in 27% (22% with ≥VGPR) in the 2.5 mg/kg group and 40% (27% with ≥VGPR) in the 3.5 mg/kg group. Median PFS was 2.1 months versus 5.8 months, respectively, and median OS was 9.4 months versus 13.8 months.
These results seem to suggest that belantamab mafodotin has similar activity in high-risk RRMM patients compared to the general RRMM population.
Following the results of DREAMM-2, belantamab mafodotin received FDA approval on 5th August 2020. Although it demonstrated an acceptable toxicity profile, belantamab mafodotin administration will be required through a risk evaluation and mitigation strategy programme due to the significant risk of ocular toxicity.55 This programme requires patients to undergo ophthalmic examinations (slit lamp and visual acuity) at baseline and prior to each dose. Even though prophylactic use of steroid eye drops seemed to provide no benefit to prevent the development of corneal AE in DREAMM-2,9,56,57 patients were advised to use preservative-free lubricant eye drops during treatment. General management of corneal toxicity consists of dose modifications and interruptions in therapy as described in the DREAMM trials, with the median time to keratopathy resolution being 2 months and visual acuity changes at 22 days.
Resistance Mechanisms
Anti-BCMA therapeutic efficacy as well as DOR may be contingent on the levels of soluble BCMA (sBCMA). This formation occurs as a result of BCMA shedding from the cell surface because of the direct cleavage by ubiquitous γ-secretase.58 It has been previously described that sBCMA prevent anti-BCMA antibody activity through competitive binding and serving as a soluble decoy for antibodies.59 Clinically, increased blood levels of sBCMA have been associated with progression of disease and shorter OS.47 The use of γ-secretase inhibitors to overcome resistance and increase anti-BCMA activity has been previously described in CAR-T therapy in vivo60 and in combination with belantamab mafodotin in vitro.61 A Phase I/II trial will evaluate the efficacy and safety of belantamab mafodotin in combination with a γ-secretase inhibitor, nirogacestat (DREAMM-5).62
Another way to combat resistance to anti-BCMA treatment is to design therapeutic agents with preferential binding affinity to membrane-bound BCMA versus sBCMA. MEDI2228 is a fully human ADC connected to a pyrrolobenzodiazepine payload, which induces DNA damage and apoptosis following internalisation. Preclinical data suggest that MEDI2228 may display weak binding to sBCMA compared to membrane-bound BCMA, as well as more potent activity to prevent MM cell proliferation and survival compared with MMAF.63,64 A Phase I study is currently enrolling patients with RRMM to evaluate the dosing and safety of MEDI2228.65
FUTURE DIRECTIONS
Due to its significant single-agent activity in MM, belantamab mafodotin is being evaluated in various combination regimens as part of a series of DREAMM trials. Two Phase I/II trials will assess the combination of belantamab mafodotin with pomalidomide and dexamethasone66 or pembrolizumab (DREAMM-4).67 Early results of a Phase I/II trial evaluating belantamab mafodotin in combination with dexamethasone and bortezomib or lenalidomide reported 78% ORR and 50% at least VGPR in a bortezomib-containing arm (DREAMM-6).68 Bortezomib and dexamethasone in combination with belantamab mafodotin or daratumumab will be studied in a Phase III trial (DREAMM-7).69 Another Phase III trial will assess belantamab mafodotin, pomalidomide, and dexamethasone combination versus bortezomib, pomalidomide, and dexamethasone (DREAMM-8).70 Finally, belantamab mafodotin in combination with triplet therapy consisting of bortezomib, lenalidomide, and dexamethasone will be compared with triplet therapy alone in a Phase III study of transplant ineligible NDMM patients (DREAMM-9).71
With multiple anti-BCMA treatment options being evaluated and nearing FDA approval, including CAR-T cell therapy and T-cell engaging bispecific antibodies, one of the most important questions will be the choice of specific anti-BCMA treatment. ADC, such as belantamab mafodotin, may provide an advantage due to its relatively simplified manufacturing process and immediate availability compared to CAR-T. In addition, ADC may be better suited in those patients who are frail as it carries no risk of cytokine release syndrome seen with other anti-BCMA treatment modalities. One of the other considerations is that BCMA expression may be preserved at disease progression allowing for different anti-BCMA therapy following relapse.72 At this time, specific successive treatment patterns for various BCMA-targeted therapies remains to be elucidated.
CONCLUSION
Evolution of MM treatment has rapidly expanded over the past 5 years, offering unique biologic targeting focussing on immunotherapeutic mechanisms to control disease progression and providing deeper and durable responses. While the most effective treatment sequence for both newly diagnosed and refractory patients has yet to be determined, these two agents offer additional therapeutic options for tailoring patient care. Strategies for selection of therapies based upon cytogenetic risk will certainly be a major driver in the MM treatment landscape and both isatuximab and belantamab mafodotin are currently being investigated in this arena. Unique triple and quadruple combinations with BCMA and CD38 moAb may offer transplant-ineligible patients desirable outcomes with a lower risk of disease progression and improved OS without compromising tolerability. In transplant-eligible patients, the ability to achieve a better disease control may allow for improved transplant-related outcomes. Finally, the approval of these agents has offered an additional lifeline to RRMM patients.





