Abstract
Paroxysmal nocturnal haemoglobinuria (PNH) is a rare, nonmalignant, haematopoietic clonal disorder that manifests with haemolytic anaemia, thrombosis, and peripheral blood cytopenias. The diagnosis is based on laboratory findings of intravascular haemolysis and flow cytometry. Clinical findings in PNH include haemolytic anaemia, thrombosis in atypical sites, or nonspecific symptoms attributable to the consequences of haemolysis. Thrombosis is the leading cause of death in PNH. Terminal complement pathway inhibition with eculizumab controls most of the symptoms of haemolysis and the life-threatening complications of PNH. However, there is still no consensus about haematopoietic stem cell transplantation (HSCT) in the management of PNH; it is the only potentially curative therapy for PNH. There are limited data and few case series about both the long-term outcomes of HSCT for PNH and the impacts of conditioning regimens on PNH clones. The authors have reviewed the findings of these studies which report on HSCT for the treatment of PNH.
INTRODUCTION
Paroxysmal nocturnal haemoglobinuria (PNH) is a rare disease of nonmalignant clonal haematopoietic stem cells with an estimated prevalence of 1–16 cases per million, which may be underestimated because of undiagnosed patients. It affects males and females almost equally.1 In 1882, for the first time, a 29-year-old man with fatigue, haemoglobinuria, and abdominal pain was described with PNH.2 PNH is an acquired disorder, characterised by life-threatening complement-mediated intravascular haemolysis, thrombosis, and bone marrow failure (BMF).3-5 Although it is known as a kind of haemolytic anaemia, the abnormality of PNH could not be explained by red blood cells.6 Treatment of PNH is still a challenge in this century because treatment options inhibit terminal complement pathway activation, ameliorate haemolysis, and thrombosis, though they are unable to manage BMF.7 Eculizumab, a first-in-class monoclonal antibody that inhibits the terminal complement pathway, is the treatment of choice for patients with severe manifestations of PNH.8 Haematopoietic stem cell transplantation (HSCT) remains the only cure for PNH but should be reserved for selected patients with suboptimal response to eculizumab because of its significantly high morbidity and mortality rates.8,9 In this manuscript, the authors compose an overview on PNH and the outcomes of HSCT in the management of PNH.
PAROXYSMAL NOCTURNAL HAEMOGLOBINURIA CLINICAL PRESENTATIONS
Clinical features of PNH are wide-ranging (Table 1) and are classified into three different forms according to flow cytometric findings, laboratory abnormalities, and bone marrow analysis: classical PNH, PNH associated with another BMF syndrome (aplastic anaemia [AA], myelodysplastic syndrome), and subclinical PNH.
Haemolysis patients can present with dyspnoea and chest or abdominal pain because of complement-mediated haemolysis which is the main mechanism of PNH.10 Periods of haemolysis can be triggered by consequences of complement activation such as infection, surgery, strenuous activity, and alcohol intake.11 Anaemia can be seen because of the combination of haemolysis and BMF.
Thrombosis is a major cause of morbidity and mortality in PNH.12 Thrombotic events occur in 40% of patients with PNH and 5–40% of patients can present with thrombosis as an initial symptom.13,14 It is noteworthy that the prevalence of thromboembolic events within the PNH population can be variable and some patients never experience thrombosis in their life.15 The exact physiopathology of the hypercoagulable state in PNH is unknown, but current data suggests that it has a multifactorial cause.13
Abdominal pain, dysphagia, and chest pain are thought to be caused by the depletion of nitric oxide (NO) because of the accumulation of high levels of free haemoglobin released into the circulation during episodes of haemolysis.10
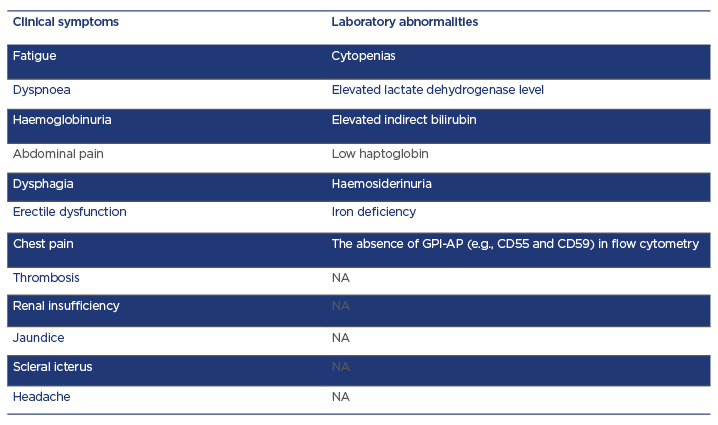
Table 1: Clinical signs and symptoms of paroxysmal nocturnal haemoglobinuria.
CD: cluster of differentiation; GPI-AP: of glycosylphosphatidylinositol anchor proteins; NA: none available.
NO depletion causes the majority of symptoms in patients with PNH.16,17 NO relaxes smooth muscle; its depletion results in excessive smooth muscle contraction and is thought to be correlated to PNH clonal size.16 Smooth muscle dystonia is responsible for dysphagia and abdominal pain.11 Pressure in the chest and dysphagia because of oesophageal spasm is very common during the haemoglobinuria episodes.18
Pulmonary hypertension is thought to occur because of NO depletion in pulmonary circulation and/or thrombosis in the pulmonary system.18 Cytopenias, in addition to complement-mediated intravascular haemolysis, are an element of BMF. BMF can overlap with PNH in patients, and PNH frequently arises in association with a defined BMF process, particularly AA and, to a lesser extent, low-grade myelodysplastic syndrome, in 50–60% and 15–20% of cases, respectively.6 Cytopenia dominates the clinical feature rather than haemolysis in patients with PNH with BMF and usually the clone size of PNH is small in these patients.5,19 Less intravascular haemolysis, low reticulocyte count, low lactic acid dehydrogenase (LDH) value, and more severe pancytopenia are expected because of the hypocellular bone marrow.20Although PNH is a nonmalignant clonal stem cell disease, progression to acute leukemia has been occasionally reported as 2–3%, which is similar to AA.21
SCREENING AND DIAGNOSIS OF PAROXYSMAL NOCTURNAL HAEMOGLOBINURIA
The main laboratory findings in PNH can be described as nonimmune haemolytic anaemia, meaning mild-to-severe anaemia, elevated serum LDH, and reticulocytosis with negative Coombs test.10 Increased unconjugated bilirubinemia and mild jaundice can also be observed.1 The occurrence of dark urine and urinary haemosiderin, both evident in intravascular haemolysis, strongly suggest PNH.22 Cytopenias and anaemia with either leukopenia and/or thrombocytopenia could be present. Even if patients have AA, PNH clones should be considered because of the overlap of these disorders, as previously mentioned. The cases with unexplained cytopenia include anaemia, nonhaemolytic anaemia, and hypoplastic myelodysplastic syndrome, which all require screening for PNH.23 Patients who are aged <50 years old; have thromboembolic events of unknown origin, especially at unexpected sites such as intra-abdominal, cerebral, and cutaneous veins; and who have cytopenias and haemolysis should be screened for PNH by flow cytometry.1,22
PNH has been shown to occur because of an acquired mutation of the PIG-A gene, which is responsible for the first step in the synthesis of glycosylphosphatidylinositol anchor proteins (GPI-AP) on the cell surface.24 Cluster of differentiation (CD)55 and CD59 are GPI-anchored proteins that block complement activation; intravascular haemolysis is blocked by CD59, and extravascular haemolysis by CD55.10 Complement-mediated haemolysis occurs in patients with PNH because these complement inhibitors are missing.25 Lack of GPI-AP on cells can be identified in peripheral blood by using flow cytometry and should detect the absence or severe deficiency of GPI-AP on at least two lineages.16,22 With flow cytometry detection of GPI-AP, deficient red blood cells could be demonstrated and categorised into Type I (normal), Type II (partial deficiency of GPI-AP), and Type III (complete deficiency of GPI-AP), which provide an estimation of the haemolysis severity.3 However, it was recognised that both haemolysis and transfusion would underestimate the clone size derived from red blood cell analyses and therefore, analysis of deficient CD55 and CD59 was also applied to granulocytes, which have a normal lifespan.10 Flow cytometry is the gold standard for screening PNH but in last few years the antibody panel that is combined with fluorescein-labeled proaerolysin, an inactive variant of bacterial aerolysin which specifically binds to GPI-AP, has shown to be particularly sensitive for PNH diagnosis and its binding is not influenced by the maturational stage of target cells.22
PAROXYSMAL NOCTURNAL HAEMOGLOBINURIA TREATMENT
Historically, management of classic PNH was based on supportive care such as transfusions of blood cells, steroids, and folate supplements.26 At present, terminal complement pathway inhibition and HSCT are the only effective and approved therapies for patients with PNH.27 Not all patients require treatment and in cases, the PNH clone may disappear spontaneously and spontaneous remission of the disease has been reported.28,29 However, if treatment is needed, the type of therapy depends on the clinical form of the disease: subclinical PNH, PNH in the setting of another BMF syndrome, or classic PNH (see Figure 1).
In subclinical PNH, patients are diagnosed when they are tested for another condition, usually cytopenia. There is no clinical or biochemical evidence of intravascular haemolysis but evidence of a concomitant BMF syndrome can often be seen in patients with subclinical PNH.6 These patients should be treated according to underlying haematologic disease.30However, it is necessary to closely monitor these patients because an expansion of the PNH clone or the cells with a PNH phenotype may occur and patients can develop classic PNH.28,30
When the patients with BMF have mild or minimal haemolysis and PNH clone at the same time, it is classified as PNH/BMF. Although variable, the percentage of GPI-AP–deficient PMN is usually relatively small (<50%) in these patients.6 The treatment should be based on underlying BMF syndrome and immunosuppressive therapy and/or HSCT may be needed for these patients.31 Moreover, eculizumab could be used concurrently with immunosupressive therapy in patients with PNH/AA.32
In classical PNH, patients usually have a large clone (>50%). Eculizumab is a U.S. Food and Drug Administration (FDA)-approved humanised monoclonal antibody that inhibits the terminal complement cascade and is highly effective in treating these patients by causing a rapid and sustained reduction of intravascular haemolysis and markedly improving quality of life for most PNH patients.7,33-35 Eculizumab binds to the complement component C5 and prevents its cleavage to C5a and C5b, which is required for formation of the membrane attack complex and reduces complement-mediated haemolysis.35
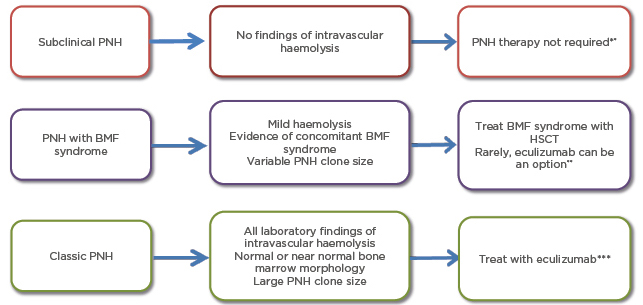
Figure 1: Clinical features and management of paroxysmal nocturnal haemoglobinuria.
*May need immunosupressive therapy if overlapping with BMF syndrome
**If patients have large PNH clone size they may benefit from eculizumab therapy
***HSCT, transfusion, erythropoietin, steroids, and splenectomy could be different treatment options. Patients who have extravascular haemolysis may especially benefit from splenectomy
BMF: bone marrow failure; HSCT: haematopoetic stem cell transplantation; PNH: paroxysmal nocturnal hemoglobinuria.
Following treatment with eculizumab, increased haemoglobin levels, transfusion independence, and normal LDH levels can be seen, and all symptoms related to haemolysis such as abdominal pain, malaise, lethargy, and dysphagia are completely resolved.36 Even though it is not possible to stop intravascular haemolysis in PNH patients with eculizumab, transfusion is an option to control anaemia.37,38 There is no cut-off haemoglobin level and it depends on the patients symptoms.15 Unlike other transfusion-dependent patients, patients with PNH have a low risk of iron overload because of the large iron loss associated with haemoglobinuria and haemosiderinuria, and these patients usually do not need any iron chelation therapy.37
Second generation complement inhibitors like ravulizumab, crovalimab, tesidolumab have been developed in the last decade.39 These new agents are promising to prolong the half-life, change the administration (oral or subcutaneous instead of intravenous), and inhibit C5 efficiently.39 Ravulizumab is similar to eculizumab in that it binds the C5 complement fraction, but conversely it has a terminal half-life that is longer by about 4-fold.40 In Phase III trials, ravulizumab was found to be as effective as eculizumab as measured by increased transfusion avoidance and LDH and haemoglobin normalisation.40 Headache was the most frequent adverse event (26.8% with ravulizumab versus 17.3% with eculizumab) but no meningococcal infections or discontinuations because of adverse events occurred.40 Crovalimab that is a long-acting anti-C5 monoclonal antibody that utilises the same mechanism as eculizumab and ravulizumab but targets a different epitope on C5.39,41,42 Although vasculitis-like symptoms may have occurred, the drug was generally well tolerated and has promising results.41,42 Tesidolumab is an another human monoclonal antibody directed against C5.43 Trials of tesidolumab aimed to overcome eculizumab resistance because of the R885H C5 polymorphism, frequently found in the Japanese population.39
PAROXYSMAL NOCTURNAL HAEMOGLOBINURIA AND HAEMATOPOIETIC STEM CELL TRANSPLANTATION
Besides the success of eculizumab as treatment for unregulated activity of the complement system, it is an expensive drug and the patient will need doses indefinitely every 2 weeks because of its short life time (median: 11.3±3.4 days).44It is a challenge for patients with PNH to be treated with a drug intravenously every other week for a lifetime.35 Despite eculizumab improving the laboratory measurements and symptoms of PNH, it is not curative and it cannot eliminate the PNH clone, meaning patients have to benefit from it as soon as they start treatment.36,45 Allogeneic HSCT remains the potential curative option for PNH through elimination of the defective clone with conditioning intensity and/or the graft-versus-haematopoiesis effect and replacement with a donor haematopoietic system.25 However HSCT for classical PNH alone is not advised in countries where eculizumab is available because of the high treatment-related mortality, though HSCT is a therapeutic option for the patients with PNH/BMF.32,46
Various centres have described their experience with HSCT for PNH and the authors analysed the studies which reported patients with PNH who underwent allogeneic HSCT (Table 2). Most studies are conducted in China, where eculizumab is not available, so usually steroid and allogeneic HSCT is considered as a first-line therapy for both classical PNH and PNH/BMF. To date, treatment with eculizumab has never been directly compared to HSCT in a randomised trial of patients with PNH.
WHICH DONOR TYPE AND CONDITIONING REGIMEN SHOULD BE PREFERRED?
Several reports of patients with PNH undergoing HSCT with a syngeneic donor, meaning the donor is an identical twin of the patient, with or without a conditioning regimen, have been published since 1979.47,56-60
Table 2: Allogeneic stem cell transplantation studies for paroxysmal nocturnal haemoglobinuria treatment.
Doukas et al.57 reported the case of a 24-year-old patient who was successfully treated with HSCT with a syngeneic donor after a myeloablative conditioning regimen and the patient remained in remission over 12 years after transplantation, with normal levels of CD59.57 Additionally, some cases have reported that a conditioning regimen has better outcomes for syngeneic donors in PNH,54,57 and syngeneic HSCT has been performed in limited number of patients with PNH.
There is not much information available currently about comparing the full matched donor types, but in two studies it has been reported that donor types matched to siblings versus unrelated matches (overall survival rates [OS]: 92.7% and 89%, respectively)49 led to similar results.46
Saso et al.54 showed poorer outcomes with alternative donor sources because of graft failure; however, it should be considered that since the time of this study, haploidentical transplantation, meaning a donor is a family member who is not human leukocyte antigen (HLA) full matched, has been developed, which has improved the management of infections and graft versus host disease (GVHD) complications.61 New, targeted therapies for GVHD and new treatments for infectious complications have been reported in the last decade.62 Some studies have reported haploidentical transplantation is as successful as full matched donors for PNH.48,50 Brodsky et al.63 reported three patients with PNH treated with haploidentical HSCT. Two had improved long-term survival but the third died from graft failure.63 These three patients did not receive eculizumab therapy and it is noteworthy that the authors clearly announced HSCT would be considered for the patients who had life-threatening thrombosis in this trial.63 Another, larger study analysed 25 patients with PNH treated with HSCT from haploidentical donors and reported that OS rate was 86.5%±7.3% 3 years after transplantation. They found similar outcomes and transplant-related complications between matched sibling donors and haploidentical donors.48 Tian et al.50 showed no differences between full matched donors and haploidentical donors in 18 cases at a median follow-up period of 20 months (range: 14–85 months), and only one patient who underwent haploidentical HSCT died from cytomegalovirus infection.
However, there are no randomised controlled trials that compare the conditioning regimen in PNH. Santarone et al.52 reported no significant difference in the rate of engraftment for patients who underwent transplantation following myeloablative conditioning compared to patients who received reduced intensity conditioning. Cooper et al.44 observed that the choice of conditioning intensity (myeloablative versus reduced-intensity) had no effect on survival rates in PNH and they found no difference in long-term survival after HSCT between patients with classical PNH and the patients who had a PNH clone associated with another marrow disorder.44 Reduced intensity conditioning regimens can eradicate PNH clones successfully and the long-term results are adequate and similar to severe AA without PNH clone with the same conditioning regimen.63-65 Considering the relatively benign and chronic nature of PNH, it seems to be more reasonable to use a reduced intensity conditioning regimen, known to be less toxic, when performing HSCT for PNH treatment.66 Transplantation from T-cell depleted stem cells from matched unrelated donors with a myeloablative conditioning regimen and a short course of eculizumab was reported as safe and an efficient donor source for young patients with PNH in one study, although there was a small case number.67
It is expected that the PNH clone will disappear after HSCT and serial measurements of the PNH cell population in both the granulocyte and monocyte lineages post-HSCT should be performed.49 However, the time can be variable from 28 days to 2 months after HSCT, depending on the conditioning regimen and donor type.44,49
What Are the Treatment-Related Complications, Mortality Rates, and Causes of Mortality?
OS rates have been reported in the range of 56–100% in multiple different studies.9,31,44,46-52,54 Previous studies have shown worse outcomes, high treatment related mortality, and graft rejection. Since these studies there has been much improvement of HSCT outcomes which explains the differences in OS rates between these trials. Being heavily transfused, the high incidence of HLA alloimmunisation, comorbidities, and PNH-related morbidities affect the outcomes of HSCT directly.53
The largest study, from the European Society for Blood and Marrow Transplantation (EBMT) in 2012, examined outcomes from 211 patients with PNH who underwent HSCT between 1978 and 2007 and compared them to outcomes in a cohort of 402 controls (patients with PNH who did not undergo HSCT between 1950 and 2005).46 In this study only one patient was documented to have a recurrence of a PNH clone after HSCT, suggesting it is a viable and curative treatment for PNH. However, the risks of HSCT are well known; the most common treatment-related complication and cause of death was GVHD, which usually occurred within the first few months of the studies.9,46,49,51 It has been shown that a history of thrombosis is an important factor for poor HSCT outcomes. Fumani et al.51 reported that the presence of thrombosis was associated with a higher mortality rate. At the time of transplantation, absolute neutrophil count >1×109/L and haemoglobin level >9 g/dL were found to be associated with better outcomes.9 The comparison between matched transplanted and nontransplanted patients with PNH in cases of thrombosis showed a poorer OS for the transplanted patients in a large study.46
Does Eculizumab Therapy Before and After Haematopoietic Stem Cell Transplantation Affect the Outcomes?
The blockage of the terminal complement pathway by eculizumab has the potential to improve or adversely affect transplant outcomes. The role of eculizumab in the transplant setting is as yet unclear. For patients with AA even with PNH clones, HSCT is still the main therapeutic option and anti-thymocyte globulin (ATG) is commonly preferred in conditioning regimens to promote engraftment and reduce GVHD, which has an effect on the complement system. However, ATG is a challenge for patients with PNH/AA because the interaction between ATG and eculizumab is still unknown.68 Patients with AA and PNH who received eculizumab as bridging therapy before HSCT have been evaluated in several studies.69,70 Using eculizumab prior to transplant does not appear to delay engraftment and there is no increased risk of infections from encapsulated organisms compared to patients with AA who underwent HSCT with no reason of PNH.69Further, eculizumab is suggested for patients with large clone size to reduce the risk of withdrawal haemolysis during and in early post-HSCT.69-71 Although in retrospective studies eculizumab was used until engraftment, it is suggested that the last dose of eculizumab should be given during the conditioning regimen because of the risk of relapse or graft failure.70 It is noteworthy that both PNH and AA are rare diseases for which transplant is increasingly utilised, but it is unlikely that there is no comparison with the patients with PNH/AA who did not receive eculizumab in previous studies and there has been no prospective trial to evaluate the role for eculizumab in HSCT.
When and Who Should Physicians Consider Haematopoietic Stem Cell Transplantation for Paroxysmal Nocturnal Haemoglobinuria Treatment?
Although there have been many improvements in HSCT outcomes in the last decade because of high-resolution HLA typing for donor selection, development of conditioning regimens, and advances in supportive care, the risks of HSCT are well known and include treatment-related mortality and GVHD.72Despite the fact that HSCT is the only curative treatment for PNH because of its ability to eliminate PNH clones, it is not recommended for patients with classical PNH.32 The decision should be given case by case and restricted to patients with BMF syndrome. It should be considered for patients who have life-threatening thrombotic disease, although thrombosis could be seen because of HSCT and transfusion dependent haemolytic anaemia.
To date, there is no prospective, intent-to-treat study comparing HSCT to eculizumab in the management of patients with PNH. Most studies have suggested that patients with PNH/BMF would benefit from HSCT. Eculizumab is efficacious in preventing thrombotic events and treating intravascular haemolysis and related manifestations in transfusion-dependent or symptomatic patients, but some patients are not willing to have lifelong treatment and a persistent need for transfusion. Additionally, it is not able to eradicate PNH clones. HSCT remains the only potential treatment for PNH, with additional implications for patients with BMF and those who are refractory to eculizumab therapy. The time from diagnosis to HSCT is also an important consideration for the patient. Both patients with PNH/BMF and classical PNH who are refractory to eculizumab, and especially young patients with severe manifestations of PNH such as severe cytopenia and thrombosis and who have a matched donor, are considered the best candidates for HSCT.
CONCLUSIONS
PNH is a clonal but nonmalignant disease that is very rare, and large case series are limited. The clinical findings and symptoms can be variable, and it can take a long time to diagnose because of physician unawareness. Eculizumab is a good treatment option for improving symptoms of intravascular haemolysis but enhanced understanding of the PNH thrombosis mechanism is needed to improve management. In the future, novel inhibitors of the alternative complement pathway could be used to improve survival and quality of life for patients with PNH. Yet, there is no therapy other than HSCT that can eradicate PNH clones. All the current drugs are useful during therapy and patients do not have the opportunity to give up treatment. Although HSCT is the only curative treatment for these patients, cases should be selected carefully because of high morbidity and mortality rates. It should be noted that the difference between OS rates, progression free survival, and quality of life between patients on lifetime treatment of eculizumab compared to patients who have undergone HSCT is still unknown.