
Abstract
Hypereosinophilic syndromes are a group of disorders characterised by significant eosinophilia and organ damage. They have proven challenging to define, diagnose, and study for many years, due in part to their variable clinical presentations, the overlap between neoplastic and reactive eosinophilia, and the lack of a universal marker of eosinophil clonality. Herein, we give an overview of the term and discuss aetiology and our approach to diagnosis.
EOSINOPHILS
Eosinophils are mature granulocytes derived from the bone marrow myeloid progenitor cells under the influence of a number of cytokines, particularly interleukin (IL)-5, though they may also mature in other organs. They are morphologically identifiable by the presence of orange granules when stained with the acidic dye, eosin. They represent approximately 1–5% of peripheral blood leukocytes (reference range: 0.05–0.5×109/L) and account for a similar fraction of nucleated cells within the bone marrow. The majority of eosinophils reside in tissue, primarily the thymus, lymph nodes, spleen, gastrointestinal tract, and uterus. Reference ranges for eosinophil numbers in these organs are less well-described.
Eosinophil granules contain many active compounds. Some (e.g. major basic protein, eosinophil cationic protein) exert direct cytotoxic effects. Others, including cytokines and chemokines, recruit and activate other cells including lymphocytes, mast cells, and fibroblasts.1 When appropriately targeted, granule release helps control pathogens, while inappropriate release has been implicated in the development of allergic inflammation, fibrosis, vasculitis, and thrombosis.
HYPEREOSINOPHILIC SYNDROMES
Terminology around these disorders has undergone many changes and is frequently confusing. The term ‘hypereosinophilic syndrome’ was first proposed in 1968 to describe disorders with persistent eosinophilia and tissue damage due to eosinophil infiltration.2 The definition was later refined to describe peripheral blood eosinophilia of unknown origin, exceeding 1.5×109/L, and persisting for at least 6 months, that was responsible for organ dysfunction/damage.3
A 2011 consensus panel proposal4 distinguishes between hypereosinophilia (HE) and hypereosinophilic syndromes (HES). Under this proposal, the term ‘hypereosinophilia’ should be used when there is peripheral blood eosinophilia of >1.5×109/L, on two occasions, at least 1 month apart, or in the presence of significant tissue eosinophilia. Identification of this latter finding is recognised as being challenging given the lack of well-established reference ranges. Finally, it acknowledges that, although blood eosinophilia usually accompanies tissue eosinophilia, tissue findings may occur in isolation.
The same panel proposes the term ‘hypereosinophilic syndrome’ be reserved for cases that fulfil the definition of HE and have otherwise unexplained organ dysfunction/damage. This damage may be due to direct cytotoxic effects of eosinophil granule contents, or occur secondary to recruitment of other cell types. The time requirement for diagnosis may be waived in cases of evolving and life-threatening organ damage in order to facilitate appropriate therapy.
HES may be classified into several broad groups. Primary (neoplastic) and secondary (reactive) HES are discussed below. HE of uncertain significance exists when neither primary nor secondary causes are apparent despite extensive investigation. Familial HE has also been described5 and appears to be due to abnormal dysregulation of cytokine signalling.6,7
Primary (Neoplastic) Hypereosinophilic Syndromes
Primary (neoplastic) HES are defined by the presence of neoplastic (clonal) eosinophils. In the 2016 classification system, the World Health Organization (WHO) recognises two key groups of eosinophilic disorders. The first, myeloid/lymphoid neoplasms with eosinophilia and rearrangements of PDGFRA, PDGFRB, or FGFR1, now includes the provisional entity with PCM1-JAK2 rearrangement.8 In order to diagnose one of these entities, the specific genetic rearrangement must be demonstrated. The second diagnostic category, chronic eosinophilic leukaemia-not otherwise specified (CEL-NOS), is categorised within the myeloproliferative neoplasms.
Clonal eosinophils may be identified in other haematological malignancies. Core binding factor acute myeloid leukaemia (AML), particularly cases with CBFB-MYH11 fusion, are associated with atypical eosinophil proliferation. The diagnostic genetic rearrangement has been demonstrated in eosinophils in both CBFB-MYH11 AML9 and in RUNX1-RUNX1T1 AML.10 In these cases, the diagnosis of AML takes precedence.
Secondary (Reactive) Hypereosinophilic Syndromes
Secondary (reactive) HES, in contrast, generally result from increased eosinophil production and may occur in a variety of disease conditions. The eosinophils are not clonally related and the eosinophilia is generally considered to be driven by cytokine/growth factor production. Infection is the most common cause of persistent eosinophilia worldwide.11 Parasite infection is particularly prevalent in developing countries and in certain areas of developed countries. Eosinophilia in these conditions tends to be most marked when parasites or their products encounter immune effector cells in tissue; purely intraluminal parasitisation is less associated with persistent eosinophilia.12
Allergic/atopic/autoimmune
Persistent eosinophilia is commonly found in allergic and atopic disease. Its development is driven by activated Type 2 T helper cells that produce eosinophil-stimulating cytokines.13 Autoimmune disorders, such as eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) and granulomatosis with polyangiitis (Wegener’s granulomatosis), are often associated with eosinophil infiltration of tissue. Hypersensitivity reactions, including allergic bronchopulmonary aspergillosis and the spectrum of drug reactions, may also result in HES.
Malignancy
Secondary HE associated with malignancy is most commonly connected with T cell malignancies, Hodgkin lymphoma, and B cell acute lymphoblastic leukaemia. Eosinophilia in these conditions is typically due to excessive cytokine (IL-5) production.4
Lymphocyte-variant hypereosinophilic syndrome
This variant of HES results from the abnormal proliferation of T helper cells. These cells are typically phenotypically aberrant, most commonly CD3–/CD4+,14 frequently express markers of activation, and produce large amounts of IL-5, demonstrated by testing supernatant after in vitro lymphocyte culture.15 Consensus diagnostic criteria for this condition are not yet established.16 The exact incidence of this subtype has been difficult to estimate, being a rare entity, though seems likely to account for 20–25% of otherwise unexplained cases of HES.15
Other
A number of other causes of significant eosinophilia are described. Episodic angioedema with eosinophilia (Gleich syndrome) is now recognised to be a cell cycling disorder of uncertain aetiology,17 with symptoms occurring at approximately monthly intervals. Eosinophilia is also seen in patients with immunoglobulin G4-related disease. Initially thought to be related to atopy, eosinophilia is reported in non-atopic patients with this condition.18 It is now considered likely to be related to the disease process itself.
APPROACH
HES are defined by the presence of organ dysfunction. Our first aim in assessing a patient with HE is to identify organ dysfunction as this allows appropriate classification and facilitates treatment. Our second aim is to identify the underlying cause. The clinical history and examination findings play a key role in guiding specific investigations. An overview to our approach is provided in Figure 1.
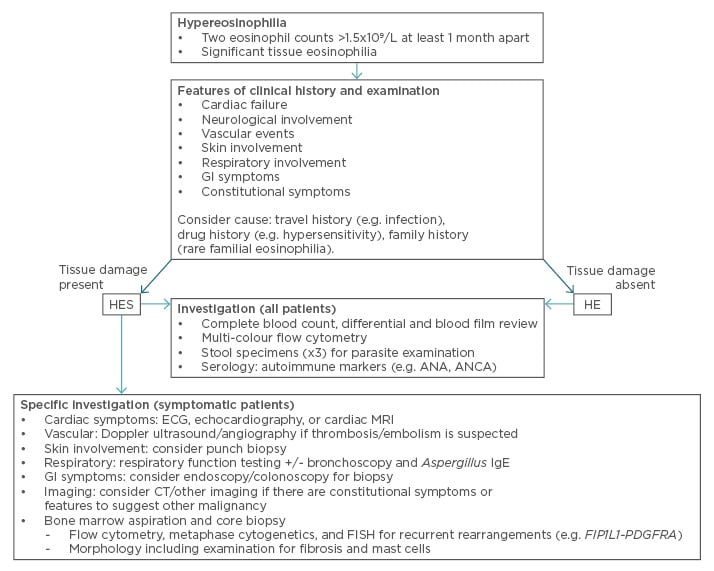
Figure 1: An overview of our approach to diagnosing HES.
ANA: anti-nuclear antibody; ANCA: anti-neutrophil cytoplasmic antibody; CT: computed tomography;
ECG: electrocardiogram; FISH: fluorescence in situ hybridisation; GI: gastrointestinal; HE: hypereosinophilia; HES: hypereosinophilic syndromes; IgE: immunoglobulin E; MRI: magnetic resonance imaging.
We specifically enquire about features of cardiac failure given the occurrence of endomyocardial fibrosis in HES. Neurological involvement may be endovascular damage or through direct activation of the clotting system, while embolic events from cardiac thrombi can present with acute arterial occlusion.
As some patients may overlook minor skin changes, particularly if long-standing, specific inquiry and examination are recommended. The nature of skin symptoms is highly variable, with some having cutaneous erythema or eczema while others may have more severe symptoms such as urticaria, angioedema, or even ulceration.
Respiratory symptoms may be prominent but are frequently non-specific. Cough and wheeze, associated with bronchospasm, may be seen while others may develop slowly progressive dyspnoea, associated with pulmonary fibrosis. Occasionally respiratory symptoms, with or without fever, may be clearly triggered by allergen exposure, and suggest hypersensitivity reactions.
A travel history, particularly to areas where parasite infection is endemic, and gastrointestinal symptoms (including abdominal pain, diarrhoea, and nausea) raise the possibility of HE secondary to parasitisation. Gut symptoms, including malabsorption, may also be seen as a direct consequence of eosinophil infiltration, cell damage, and fibrosis.
The presence of constitutional symptoms (night sweats, fever, >10% unintended weight loss) should prompt consideration of underlying malignancy. A detailed medication history is essential given the potential for hypersensitivity reactions to cause eosinophilia.
DIAGNOSTIC TESTING
We use an eosinophil count >1.5×109/L on at least two occasions 1 month apart to define HE.4 In the absence of organ damage we take a stepwise approach to diagnosis, attempting to exclude secondary causes before investigating neoplastic processes. If organ damage is identified, we typically investigate primary and secondary causes of HES simultaneously to hasten diagnosis, allow specific therapy, and minimise further tissue damage. If organ damage is acute, severe, or progressive we often commence corticosteroid therapy concurrent with diagnostic testing.19
Investigation for organ dysfunction should be guided by clinical history. In asymptomatic patients with incidental HE we typically defer such investigation. In symptomatic patients, we perform investigations targeting the involved organ system(s). These may include echocardiography, computed tomography (e.g. brain, chest, abdomen), and Doppler ultrasound to exclude vascular abnormalities. Contrast-enhanced cardiac magnetic resonance imaging has been increasingly used in the evaluation of cardiac fibrosis. It is superior to echocardiography and other imaging techniques20 and has the advantage of allowing detailed mapping of fibrosis as well as being more sensitive for the detection of ventricular thrombi. Disadvantages include the need for gadolinium contrast administration, limiting use in those with renal failure, and, in some regions, limited availability.
Tissue biopsy may be required in some cases where symptoms are partially explained by other medical conditions or where eosinophil infiltration of the affected organ is unproven. Given the challenges inherent to determining significance of eosinophil infiltration, particularly in assigning causality of organ dysfunction, discussion with the reviewing pathologist is highly recommended. Tissue biopsy may also demonstrate atypical lymphoid infiltration in cases of lymphocyte-variant HES.21
Blood counts and morphological review of the blood film are essential to diagnosis. Blood film review may identify abnormalities, including dysplastic eosinophils, to suggest primary HES. Secondary HES may also be morphologically identified with conditions such as acute lymphoblastic leukaemia and, rarely, circulating parasites are identifiable. Determination of serum tryptase is suggested given that its elevation is a minor criterion for the diagnosis of systemic mastocytosis.22 Multi-colour flow cytometry should be performed in all patients. This allows detection of aberrant T cell immunophenotypes associated with lymphocyte-variant HES. Our preference is to perform this test on bone marrow though we consider peripheral blood flow cytometry in asymptomatic patients with HE.
T cell receptor (TCR) gene rearrangement studies have been increasingly used in the setting of eosinophilia to investigate T cell clonality and lymphocyte-variant HES. Clonal rearrangements are common, with one study finding 18 out of 42 (42.8%) patients to have TCR clonality.23 Complicating matters, however, was the finding of clonal rearrangements in two patients with the FIP1L1-PDGFRA fusion product. This is in keeping with other work where clonal TCR rearrangement may be found in benign lymphoid proliferation.24 As such, the finding of a TCR rearrangement, in the absence of immunophenotypic or aberrant cytokine production, should not be considered as diagnostic.25 We recommend TCR gene rearrangement studies to confirm clonality if the diagnosis of lymphocyte-variant HES is strongly suspected; we do not recommend these studies be performed routinely in those without flow cytometric abnormalities (e.g. aberrant immunophenotype, expansion of specific lymphocyte subsets).
Microbiological investigations should be performed. Three or more fresh stool specimens for parasite examination should be obtained. Serological testing for specific parasites may be appropriate in selected cases; testing for Aspergillus-specific immunoglobulin E may be useful if allergic bronchopulmonary aspergillosis is suspected. In the presence of respiratory symptoms, we request respiratory function testing. In selected cases, particularly where a cause for eosinophilia remains unclear, we refer for bronchoscopic investigation to obtain diagnostic lavage and exclude pulmonary infection.
In patients with organ damage, those with lymphocytosis, immunophenotypic abnormalities by flow cytometry or morphological abnormalities (including circulating precursors and dysplasia) on blood film examination, or after exclusion of secondary causes, we perform bone marrow aspiration and trephine biopsy. The aspirate specimen should be submitted for morphological evaluation, metaphase cytogenetics, fluorescence in situ hybridisation, and flow cytometry. The core biopsy should be submitted for pathological review, including assessment for reticulin and collagen fibrosis. Other immunohistochemical studies may be warranted depending on findings. Specific consideration should be given to the presence of systemic mastocytosis and occult neoplasia.
High-throughput DNA sequencing does not yet have a definitive role in the diagnosis, prognostication, or identification of therapeutic targets in HES. A number of recent publications26-28 have examined the role of sequencing in these disorders, particularly in identifying clonal molecular abnormalities in idiopathic HE, though this work has yet to influence routine clinical practice. Accordingly, we do not routinely request extended DNA sequencing in our investigative approach.
Myeloid/lymphoid neoplasms with eosinophilia and recurrent genetic rearrangements should be specifically excluded. The majority of these cases have a cryptic deletion of chromosome 4q12 creating the FIP1L1-PDGFRA fusion gene.29 This cryptic deletion can be demonstrated by fluorescence in situ hybridisation studies. While PDGFRB and FGF1 are recognised to have multiple gene fusion partners, the majority of these are detectable with metaphase cytogenetics.30 The diagnosis of CEL-NOS may only be established after recurrent genetic lesions, including those associated with AML, have been excluded. CEL-NOS is otherwise defined by the presence of eosinophil clonality (shown by cytogenetic or molecular testing) or the presence of increased numbers of blast cells (>2% in blood; >5% in marrow).29
FUTURE DEVELOPMENTS
A number of important questions are, as yet, unanswered. Given the relative rarity of these diseases and previous challenges in establishing aetiology, our understanding of optimal diagnostic criteria remains based on expert opinion. International collaboration and further study may refine categorisation. Advances in diagnostic techniques may allow for direct demonstration of eosinophil clonality/polyclonality as a means of differentiating neoplastic HES from secondary/ reactive eosinophilia as well as allowing disease monitoring. While eosinophils play a role in the pathogenesis of organ dysfunction, the mechanisms by which this damage occurs are poorly understood. Further research may clarify the extent of eosinophil-mediated cytotoxicity compared to the consequences of cell recruitment. Understanding these contributions may facilitate therapy to block and reverse organ damage.
SUMMARY
HES were first defined on the basis of peripheral blood eosinophilia and tissue damage. Developments in laboratory techniques have allowed for precise aetiological classification of neoplastic eosinophilia; other techniques have highlighted the role of lymphocytes and cytokine signalling in driving eosinophilia. Ongoing developments in DNA sequencing and immunophenotyping have the potential to further classify disease and identify therapeutic targets.





