
Abstract
The emergence of B cell receptor (BCR) kinase inhibitors has recently changed the treatment landscape in chronic lymphocytic leukaemia (CLL). The inhibitors that selectively target potential kinases downstream from BCR (particularly Bruton’s tyrosine kinase [BTK] and phosphoinositide 3-kinase [PI3K]) have replaced conventional chemotherapy for high-risk CLL. Ibrutinib and idelalisib are the respective first-in-class BTK and PI3K-δ inhibitors that are US Food and Drug Administration (FDA) approved for CLL treatment, with promising second-generation molecules under development. Differing from idelalisib, duvelisib (IPI-145) inhibits both delta and gamma isoforms of PI3K. Kinase inhibitors have gained popularity in the clinic primarily due to their ability to induce remissions in the vast majority of patients, even in patients with high-risk disease features, without causing haematotoxicity. In particular, they interfere with the homing capabilities of CLL cells residing in their respective microenvironments and cause lymphocytosis via redistribution of tissue-resident CLL cells into the peripheral blood. Thereby, BCR inhibitors can seek out and target hiding CLL cells in the lymph node and marrow niches. In this review, we discuss laboratory and clinical aspects of the BCR inhibitors that have recently advanced the treatment of B cell malignancies, with a particular emphasis on CLL. Despite the excitement about this new class of compounds targeting BCR signalling, single agent therapy with kinase inhibitors has limitations, requiring continuous kinase suppression to maintain remissions, which generally are partial remissions, indicating that combination strategies will become important for moving the field forward.
RETHINKING CHRONIC LYMPHOCYTIC LEUKAEMIA BIOLOGY AND RISK FACTORS
Chronic lymphocytic leukaemia (CLL) is a B cell malignancy that is relatively common in the elderly (median age, 71 years), found preferentially in males. Negative prognostic markers for CLL include cytogenetic and molecular markers, including del17p, del11q, and unmutated immunoglobulin (Ig)VH. The clinical outcome or prognosis for patients with these markers is generally poor. Continued clinical and laboratory research has brought the treatment of CLL a long way from the original IFN-α treatments of 30 years ago. For many decades, non-targeted chemotherapeutic agents such as single agent fludarabine,1 cyclophosphamide, or both in combination,2 have been the front-line therapy for CLL patients. Over the past decade, the addition of the monoclonal antibody rituximab in combination with the purine analogue (fludarabine) and alkylator (cytoxan), known as ‘FCR’, has become a standard-of-care therapy with high efficacy, prolonged overall survival (OS), and acceptable toxicity.3 Chemo-immunotherapy has improved response rate (RR) and OS4 with a median disease-free survival of 5 years.5 Over 50% of patients with IgVH-mutated CLL remained progression-free for >8 years after treatment with FCR.6,7 However, given the quiescent nature of CLL cells, it is uncertain if non-targeted DNA damaging regimens could be of any real benefit in the targeting of resting CLL B cells. On the same note, identification of new potential targets and target inhibition through a greater understanding of CLL biology have recently transformed the landscape of treatment. In particular, the emergence of targeted therapies like B cell lymphoma-2 (Bcl-2) inhibitors8,9 (ABT-199, GDC-0199, venetoclax, AbbVie, Chicago, Illinois, USA) and B cell receptor (BCR) kinase inhibitors (ibrutinib and idelalisib)10-13 have significantly shifted the treatment paradigm for this disease.
B CELL RECEPTOR PATHWAY/B CELL RECEPTOR KINASE INHIBITORS
One broad target of B cell malignancies is those driven by BCR receptor signalling, downstream of which are many potential kinases activated in a cascade fashion, allowing them to significantly drive CLL pathophysiology. Identification of BCR kinases as potential targets for treatment and the discovery of BCR kinase inhibitors to selectively and specifically target BCR pathway kinases have been major breakthroughs in CLL therapy (Figure 1).
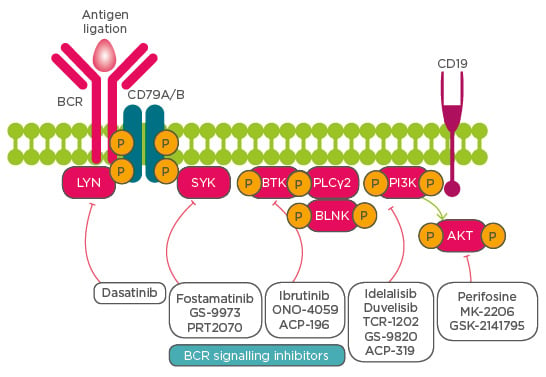
Figure 1: Schematic representing activation of various kinases downstream of the B cell receptor signalling pathway and their inhibitors for therapy of patients with chronic lymphocytic leukaemia.
BCR B cell receptor; BTK Bruton’s tyrosine kinase; SYK spleen tyrosine kinase; BLNK B cell linker; PLCγ2 phospholipase Cγ2; PI3K phosphoinositide 3-kinase.
IBRUTINIB: FIRST APPROVED BRUTON’S TYROSINE KINASE INHIBITOR
Ibrutinib (marketed as Imbruvica®) is the first- in-class, covalent, small molecule oral inhibitor of Bruton’s tyrosine kinase (BTK) inhibitor, a protein downstream of the BCR pathway essential for CLL cell survival and proliferation.10 Ibrutinib received all four FDA expedited development programmes: Fast Track designation, Breakthrough Therapy designation, Priority Review, and Accelerated Approval14 for the treatment of previously treated CLL or for the first-line therapy of high-risk CLL with del17p or TP53 mutations. The activity and safety profile of single-agent ibrutinib in CLL with del17p is encouraging, supporting a novel treatment option for patients with high-risk CLL in both first-line and second-line settings.15 Of note, ibrutinib was also well-tolerated in healthy volunteers.16
IBRUTINIB: TARGETS THE HIDING CHRONIC LYMPHOCYTIC LEUKAEMIA CELLS
Rethinking CLL as a dynamic disease has helped in viewing it differently from the perspective of therapeutics. CLL cells often depend on microenvironmental factors for proliferation and survival. In particular, tissue-resident CLL cells exhibit sustained activation of both BCR and NF-κB pathways.17 A study evaluating in vivo effects of ibrutinib on tumour cell activation and proliferation in blood, lymph node, and bone marrow compartments showed a rapid and sustained downregulation of BCR and NF-κB signalling in CLL cells from both the peripheral blood and tissue compartments and decreased tumour proliferation that was independent of prognostic factors such as IgHV status, del17p, or prior treatment history.18 Another study conducted on serial samples taken from CLL patients before and after initiation of ibrutinib reported a blockage of cell proliferation with a reduction in Ki-67 expression and bromodeoxyuridine incorporation, indicating that ibrutinib classically targets hiding CLL cells (Figure 2).19
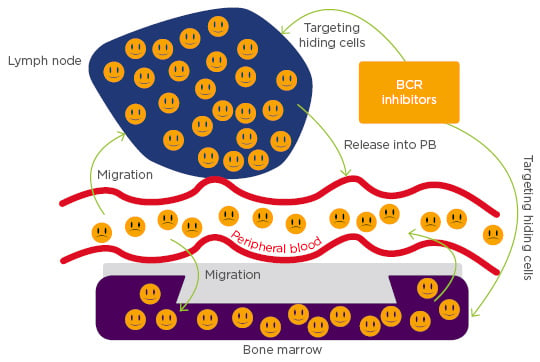
Figure 2: Chronic lymphocytic leukaemia cells migrate towards the chemokines expressed on the tissue microenvironments, such as bone marrow and/or lymph node, where they acquire resistance to conventional chemotherapy. B cell receptor inhibitors are a class of compounds that block the homing capabilities of chronic lymphocytic leukaemia cells and liberate them into the peripheral blood, where they can easily be targeted by other agents.
BCR: B cell receptor; PB: peripheral blood.
LYMPHOCYTOSIS: CLASS EFFECT OF B CELL RECEPTOR INHIBITORS
A distinct feature of BCR inhibitors is their ability to cause rapid shrinkage of the lymph nodes within days or weeks upon initiation of treatment. Of note, it was previously known that various other agents such as glucocorticoids, 7-hydroxystaurosporine (UCN-01), and rapamycin (sirolimus) cause similar effects on lymphocyte redistribution, an off-target lymphocytosis effect (as discussed in a recent review).20 The occurrence of lymphocytosis with BCR signalling inhibitors was first observed with fostamatinib (R788), an inhibitor of spleen tyrosine kinase downstream of BCR. Phase I/II studies of single-agents in patients with relapsed refractory CLL demonstrated an early lymphocytosis of >50% of baseline in 9 of 11 patients, which was associated with a reduction in lymphadenopathy, indicating that inhibition of spleen tyrosine kinase leads to increased trafficking of CLL cells out of the nodal microenvironment and into the peripheral blood where they eventually undergo apoptosis.21,22
The reduction of tumour burden with BCR signalling inhibitors was initially received with great enthusiasm by the patient community and their treating clinicians. Later, it was noted that the lymph node shrinkage is associated with a phenomenon called lymphocytosis i.e. elevation of lymphocyte count in the peripheral blood. Initial doses of ibrutinib peaked at a 66% increase in baseline absolute lymphocyte count in >40% of patients within 24 hours of treatment. Consequently, the observed increase in absolute lymphocyte count was interpreted as a sign of progressive disease and misguidedly viewed as a reason to stop the treatment. Genomic expression profiling studies indicated that the molecular signature of redistributed cells resembled that of cells originating from tissues of the microenvironment (like bone marrow and lymph nodes that serve as havens for CLL cell growth and survival). Circulating CLL cells showed an increased proliferative marker, Ki67, indicating an efflux of leukaemic cells from the tissue compartments into the blood.23
Several studies were conducted to understand the event of lymphocytosis and its associated kinetics in the redistribution of CLL cells. Before initiating therapy, the DNA in proliferating cells was labelled with deuterium (patients were asked to drink deuterated water) and the effects of ibrutinib on leukaemia cell kinetics (proliferation and death rates) and redistribution of cells from lymphoid tissues (trafficking) after ibrutinib administration were determined. The data confirmed that after initiation of ibrutinib, there was a rapid increase in radio-labelled CLL cells in the blood rather than the emergence of newly-divided unlabelled cells, indicating mobilisation of CLL cells from proliferating centres.24 A statistical modelling study conducted to characterise the dynamics of the redistribution phenomenon and pattern of treatment related lymphocytosis demonstrated no significant differences in the pattern of lymphocytosis between cohorts of treatment-naïve elderly CLL, relapsed/refractory CLL, and high-risk CLL, including no detectable dose effect.25 Another correlative study (using mathematical modelling), between serial lymphocyte counts and volumetric changes in lymph node and spleen sizes during ibrutinib therapy, reported that <2% of peripheral blood CLL cells and <3% of tissue CLL cells underwent apoptosis per day. The fraction of the tissue CLL cells redistributed into the bloodstream during therapy was estimated to be 23% of the total tissue disease burden, indicating that the reduction of tissue disease burden by ibrutinib is primarily due to apoptosis of CLL cells rather than due to their release from the nodal compartments.26
These reports clearly indicated that the BCR inhibitors are unique in causing interference between CLL cells and the associated microenvironment by disrupting the migration and homing capabilities of resident cells. It is this novel mechanism of action of the BCR inhibitors that causes reduced homing of leukocytes and CLL cell redistribution from tissue sites into the peripheral blood, contributing to peripheral lymphocytosis. Additionally, the rapid redistribution of cells leading to either a transient or a prolonged event was shown to be associated with favourable prognostic features.27
CLINICAL ACTIVITY OF IBRUTINIB
Three years of ibrutinib as a front-line regimen or salvage therapy has demonstrated excellent clinical activity in patients.28 Studies published on responses to ibrutinib monotherapy10,15,29 or in combination with rituximab,30 ofatumumab,31 rituximab and bendamustine,32,33 or chemoimmunotherapy34 have been encouraging. Comparison of ibrutinib to ofatumumab demonstrated ibrutinib as superior to ofatumumab in difficult-to-treat patients with relapsed or refractory CLL as assessed by progression-free survival (PFS), OS, and clinical response.35 In a recent study, ibrutinib was found superior to chlorambucil in previously untreated patients with CLL as assessed by PFS, OS, RR, and improvement in haematological parameters.13 A prospective, multicentre, placebo controlled, double-blind Phase III study was subsequently initiated with the objective of comparing the efficacy and safety of ibrutinib to that of a watch-and-wait approach in Binet Stage A CLL.36
RESISTANCE MECHANISMS FOR IBRUTINIB
Although ibrutinib induces durable responses in a majority of CLL patients, it also can result in patients developing therapy resistance, especially in patients with del17p and complex cytogenetic abnormalities. Mutation in a cysteine residue of the BTK protein (C481), to which ibrutinib covalently and irreversibly bonds to inhibit its kinase activity, has been described as a common resistance mechanism. Additional mutations in PLCγ2, which is an immediate downstream target of BTK, were also observed in ibrutinib-resistant patients.37 Whole- exome, deep sequencing studies conducted to dissect the evolution of ibrutinib resistance resulted in the detection of the BTK-C481S mutation, multiple PLCG2 mutations, and the expansion of clones harbouring del8p with additional driver mutations (EP300, MLL2, and EIF2A), suggesting that ibrutinib therapy favours the selection and expansion of rare sub-clones already present before treatment.38
CONTINUOUS KINASE SUPPRESSION REQUIRED
Long-term follow-up results with kinase inhibitors and historical data with ibrutinib suggested a requirement for continuous kinase suppression and other key issues, including response to ibrutinib reintroduction and effect of treatment interruption on secondary resistance to ibrutinib. At the clinical level, responses are partial, indefinite daily use of the drug is required, and the time to secondary resistance was found to increase with duration of treatment. A study comparing the baseline factors in association with discontinuation of ibrutinib reported poor prognosis and disease progression after discontinuation of therapy with associated mutations in BTK and PLCG2.39 Another study reported that a majority of patients with CLL who discontinued treatment with ibrutinib were found to have high-risk features (94% with unmutated IgVH and 58% with del17p by interphase fluorescence in situ hybridisation and 76% of these patients died after discontinuing ibrutinib within a median OS of 3.1 months after discontinuation). Most patients with RR-CLL who prematurely discontinued ibrutinib were difficult to treat and had poor outcomes.40 Despite this, at current pricing, the average wholesale price of ibrutinib-based treatment in the USA is approximately $118,000 per year and has made a substantial impact on the cumulative cost of CLL care.
IDELALISIB: A PHOSPHOINOSITIDE 3-KINASE DELTA INHIBITOR
Phosphoinositide 3-kinase (PI3K) delta expression, a component of BCR signalling, was found overexpressed in leukocytes, providing a mechanistic rationale for inhibition of the delta isoform in B cell malignancies. Idelalisib (Zydelig R GS-1101, CAL-101) is a first-in-class, targeted, oral inhibitor that selectively inhibits P110δ expression.11 Preclinical studies demonstrated that idelalisib inhibits BCR signalling and chemokine networks in CLL.41 The agent is FDA approved in combination with rituximab for the treatment of relapsed CLL.12 The combination of idelalisib and rituximab, as compared with placebo and rituximab, significantly improved PFS, RR, and OS among patients with relapsed CLL who were less able to receive chemotherapy. To evaluate idelalisib as an initial therapy, 64 treatment-naïve older patients with CLL were treated with rituximab and idelalisib twice daily continuously for 48 weeks. The median time on treatment was 22.4 months with an overall RR of 97%, and a complete response rate of 19%. Interestingly, the overall RR was 100% in patients with del17p/TP53 mutations and 97% in those with unmutated IgHV. PFS was 83% at 36 months.42-46
TOXICITIES FOR B CELL RECEPTOR SIGNALLING INHIBITORS
Despite improved PFS, RR, and OS among patients, resistance and toxicities have been two common companions for these agents as well. Gilead Sciences Inc. (Foster City, California, USA) announced that they are stopping six clinical trials exploring idelalisib in combination with other therapies due to reports of an increased rate of adverse events, especially infectious and autoimmune complications in haematological malignancies. The FDA cautioned clinicians not to prescribe the drug for patients with previously untreated CLL or in combination with other cancer medications until the cause is investigated.
DUVELISIB, PHOSPHOINOSITIDE 3-KINASE DELTA AND GAMMA INHIBITOR
Duvelisib (IPI-145), a potent PI3K inhibitor, is different from idelalisib in that it inhibits both delta/gamma isoforms of PI3K in CLL.47,48 Even in the setting of BTK C481S mutation, duvelisib proved effective at reducing downstream PI3K signalling as evidenced by diminished AKT phosphorylation.49 Additional studies indicated that combined inhibition of delta and gamma isoforms of PI3K could be beneficial as each isoform has distinct and non-overlapping functions in CLL biology and hence could enhance the suppression of malignant B cell growth, survival, and migration.50 Duvelisib is currently in Phase III clinical trials for haematological malignancies. The results from those studies will determine the future success of this class of agents.
CONCLUSIONS
Many new and effective targeted agents are competing for a role in the treatment of CLL,51 and whether second-generation molecules prove to be significantly better than the already approved agents (ibrutinib, idelalisib, venetoclax) remains to be seen. Without sufficient long-term follow-up data, it remains difficult to answer questions about the future use, action, and side effects of BCR inhibitors in a definitive fashion. As data emerge from clinical trials with highly active therapies, clinicians caring for patients are left with the question of how to best incorporate these agents into their treatment approaches. Studies combining BCR kinase inhibitors with chemo-immunotherapy (bendamustine and rituximab) demonstrated added toxicity but no clear added benefit from the chemo-immunotherapy in terms of PFS or OS, making it less likely that such combinations will move forward.32 It is very well possible BCR signalling inhibitors, such as BTK and PI3K inhibitors, will be most effective when given in conjunction with other classes of agents such as antibodies or other targeted therapies (venetoclax; inhibitor of Bcl-2, a guard-rail of apoptosis) that could eventually kill the residual CLL cells efflux to the peripheral blood. In summary, kinase inhibitors have the ability to interfere with the homing capabilities of CLL cells; monotherapy kinase inhibitors have limitations as most responses are partial remissions, suggesting that an appropriate combination of strategies will be critical in moving the field forward.52,53





