
Abstract
This case study deals with a 32-year-old Indian male patient who presented with a traumatic head injury in the hospital, experienced uncontrolled bleeding after conducting surgery, and was eventually diagnosed with Glanzmann thrombasthenia. Glanzmann thrombasthenia is a rare hereditary blood clotting disorder characterised by a lack of platelet aggregation due to the absence of platelet glycoprotein IIb/IIIa. This occurrence is generally triggered by consanguineous marriages and is apparent in approximately one in one million people. Education and raising awareness about consanguinity in communities may help to reduce challenging, unusual genetic diseases.
INTRODUCTION
Eduard Glanzmann first reported Glanzmann thrombasthenia (GT) in 1918 after he identified a functional abnormality of platelets with defective clot retraction. Glanzmann thrombasthenia is a rare autosomal recessive disorder with normal or sub-normal platelet count, prolonged bleeding time, and deficiency or absence of platelet aggregation.1,2 It is a rare genetic platelet disorder in which the platelet glycoprotein IIb/IIIa (GPIIb/IIIa) complex is affected, with an occurrence of one in one million.3 It is commonly seen in populations with an autosomal recessive pattern of inheritance with increased consanguinity. An acquired form of GT in which autoantibodies to the glycoprotein complex interfere with normal functioning has also been reported.4
Integrin αIIbβ3, formerly known as GPIIb/IIIa, is a platelet receptor for fibrinogen. It shifts to its active configuration to allow for fibrinogen binding when platelets are activated. As platelets bind the fibrinogen, they accumulate and provide primary haemostasis. Bleeding can be spontaneous or occur with an injury without functioning fibrinogen receptors or without enough of them.5,6 Platelets in GT are not as effective in producing thrombin, an integral part of converting fibrinogen to fibrin.7 When cross-linked fibrin stabilises the platelet plug, secondary haemostasis occurs and is thus also affected by GT.8
There are three groups of GTs in patients: Type I, Type II, and variations. Patients with Type I have <5% of GPIIb/IIIa; patients with Type II have 5–20% of the average amount of GPIIb/IIIa; and patients with variant types have one-half of the average quantities of GPIIb/IIIa. Type I is the most prevalent, but there is little connection between the seriousness of the disease and the subtypes. Purpura, epistaxis, gingival haemorrhage, and menorrhagia are the recurring features seen in GT, as well as excessive bleeding following dental extraction and surgery.6
CASE DESCRIPTION
A 32-year-old Indian male patient with loss of consciousness presented to the emergency department shortly after a serious traumatic head injury. He had undergone one episode of seizure at the time of the accident. There were no other comorbidities, he was a non-smoker, and had an occasional alcohol use disorder. He had no immediate family support system. Due to the head injury, the patient underwent radiological tests including a CT brain scan that revealed a right temporoparietal intracranial haematoma. The neurosurgeon immediately performed decompressive craniotomy surgery. The operation profile revealed haemoglobin 12.7 g/dL (normal range: 13–18 g/dL); blood group O; positive Rhesus typing; bleeding time of 2 minutes and 10 seconds (normal range: 1–3 minutes); clotting time of 3 minutes and 40 seconds (normal range: 3–7 minutes); random blood glucose of 162 mg/dL (normal range: 60–140 mg/dL); blood urea 29.0 mg/dL (normal ranges: 10–40 mg/dL); serum creatinine of 0.8 mg/dL (normal range: 0.6–1.5 mg/dL); negative hepatitis B surface antigen; negative for HIV-1 and -2; and negative for hepatitis C virus 1 and 2. During the surgery, there was uncontrolled bleeding at the surgical site despite all measures being taken to control the bleeding. Two pints of blood were immediately transfused. After controlling the bleeding, the haematoma was removed, and the surgical site was closed in layers.
Six units of fresh frozen plasma and two bottles of blood were post-operatively transfused, and the patient was examined for further haematological tests such as activated partial thromboplastin time (PTT; 35.7 sec); mean normal PTT (32.2 sec); prothrombin time (PT; 17.4 sec); mean normal PT (13.1 sec); and international normalised ratio (1.57). Re-examination of PT with international normalised ratio revealed 16.0 sec increase and activated PTT of 29.6. Other laboratory tests were performed on serum electrolytes: sodium (139 mmol/L); potassium (4.1 mmol/L); and chloride (99 mmol/L). A complete blood picture showed total white blood cell count of 12×109/L, a differential count of neutrophils (92%), eosinophils (1%), lymphocytes (55%), monocytes (2%), basophils (0%), haemoglobin (12.7 g/dL), total red blood cell count (4.6 million/mm3), and platelet count (194×103 /mm3).
After the surgery, repeat CT brain scan (plain) showed right-sided temporoparietal and frontal contusions with moderate mass effect, for which emergency re-exploration surgery was performed. During the emergency re-exploration, the surgeons evacuated the temporal intracranial haematoma but the frontal haematoma was deferred. At the time of surgery, there was another bout of uncontrolled bleeding that lasted for 1 hour despite compression with AbGel®©™ surgical cottonoids (Shri Gopal Krishna Labs Pvt. Ltd., Mumbai, India).
The emergency re-exploration surgery and CT cerebral angiogram revealed decreased haematoma in the right temporoparietal region with air foci within the stable size of the right frontal haematoma; stable intraventricular haemorrhage and tentorial haemorrhage; persistent mass effect over the right lateral ventricle with mild midline shift to the left (5 mm); and no evidence of aneurysm or any focal stenosis.
For the case of uncontrolled bleeding during surgery, the haematologist carried out a platelet aggregometry test (Table 1) and the results revealed a GT diagnosis. Upon diagnosis, the patient’s caregiver was thoroughly questioned and revealed that the patient was born to consanguineous parents. A plain CT brain scan revealed the right frontal, temporal, parietal decompressive craniotomy, and the right frontal contusions slowly resolved during the hospital stay. In post-operative care, the patient was treated with supportive care, intravenous fluids, antibiotics, analgesics, anticonvulsants, anti-oedema measures, proton pump inhibitors, and anti-emetics. After haemodynamic stability, the patient was discharged with a good clinical response.
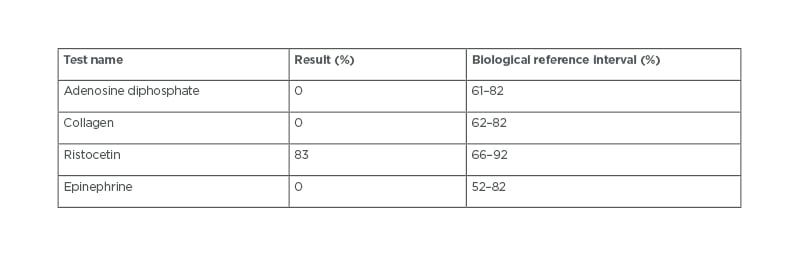
Table 1: Platelet aggregometry test.
DISCUSSION
GT is an inherited platelet function disorder and is extremely rare except in populations where consanguineous marriages are common such as in Iranian, Southern Indian, and Iraqi Jewish populations.9,10 Toogeh et al.11 conducted a retrospective descriptive study of 382 patients and concluded that most cases of GT were consanguineously born. Nelson et al.12 identified 15 unrelated patients diagnosed with GT, 11 of whom were born to consanguineous parents and six with a family history of bleeding. The authors concluded that their results confirmed the genetic diversity of mutations and polymorphisms in αIIb and β3 genes in patients with GT in Southern India and provided insight into the structure and function of relationships in these genes, but they did not identify any clear genotype or phenotype correlations. In this current study, the patient was born to consanguineous parents.
Early reports addressed the clinical heterogeneity of this bleeding syndrome: some patients had minor bruising, while others had regular, serious, and possibly fatal bleeding. Haemorrhagic symptoms occurred only in patients who were homozygous for GT-causing mutations; heterozygous diseases are often asymptomatic, although they have only a semi-normal platelet αIIbβ3 concentration.13 Zheng et al.14 explored the mechanisms underlying thrombocytopenia and the bleeding phenotype in a patient with acquired GT, concluding that platelet desialylation was mediated by anti-Fc-γ receptor IIa, independent of platelet activation, in a patient with acquired GT.
In most cases, there were rapid clinical signs of bleeding after birth, even though GT is rarely diagnosed later in life. Epistaxis is a common cause of serious bleeding and is usually more severe in childhood.15 Gopalakrishnan et al.16 reported a case on medical and surgical management of an 11-year-old female diagnosed with GT who had undergone maxillary cyst enucleation. They concluded patients diagnosed with GT show signs of bleeding very early in life.
Duncan et al.17 conducted research on GT to identify the burden of disease for patients and caregivers through a better understanding of the management and psychosocial impact of this disorder, concluding that most patients with GT are diagnosed early, with >50% of participants diagnosed with GT within 1 year.
Much of the literature has concluded that GT is diagnosed early, in contrast to this study in which the patient had GT newly diagnosed during surgery at the age of 32.
Cherian et al.18 detailed a 28-year-old male with GT who received a late diagnosis. Swathi et al.19 identified an 18-year-old female who had a recent diagnosis of GT but had a history of prolonged bleeding after mild trauma, a propensity to bleed, including purpura in areas of easy bruising, gum bleeding, and prolonged bleeding time after abrasions and insect stings.
GT, like many other bleeding disorders, has a multi-tiered treatment structure. Initial care for minor bleeding episodes may include local pressure, cauterisation, sutures, or ice therapy.20 Platelet transfusion is routine for GT as surgical prophylaxis and treatment for moderate to serious bleeding. Many patients benefit from preventative care and symptomatic treatment. Curative treatment with haematopoietic stem cell transplant has been effective in a limited number of patients with GT who have an extremely poor quality of life. Family planning discussions, which include the possibility of childbirth and the children who will be affected, are also important for some patients.7,8
CONCLUSION
This case report highlights a case of newly diagnosed GT at 32 years of age, during surgery. GT is a rare hereditary bleeding disorder caused mainly by consanguineous marriages. Education and creating awareness of these marriages is a good preventive method as they are at higher risk of rare genetic disorders.





