Abstract
Chimeric antigen receptor (CAR) T cells have emerged as an effective therapy for patients with relapsed and refractory haematological malignancies. However, there are many challenges preventing clinical efficacy and thus broader translation of this approach. These hurdles include poor autologous T cell fitness, manufacturing issues and lack of conserved tumour-restricted antigens to target. Recent efforts have been directed toward incorporating genome editing technologies to address these challenges and develop potent CAR T cell therapies for a diverse array of haematopoietic cancers. In this review, the authors discuss gene editing strategies that have been employed to augment CAR T cell fitness, generate allogeneic ‘off-the-shelf’ CAR T cell products, and safely target elusive myeloid and T cell cancers that often lack appropriate tumour-specific antigens.
INTRODUCTION
Haematologic malignancies are a heterogeneous group of cancers, which include leukaemia, lymphoma, and multiple myeloma. These diseases are a major global health burden, with an estimated 1.28 million new cases annually, representing approximately 6–7% of all global cancer cases.1 Incidence of these malignancies have been on the rise, with a 26% and 45% increase in leukaemia and non-Hodgkin lymphoma, respectively, from 2006 to 2016.2 Current standard treatments for haematologic malignancies include stem cell transplantation, chemotherapy, and radiation therapy. These therapies have traditionally focused on reducing the burden of malignant cells, and an expanding palette of drugs targeting these tumour cells is now clinically available. Although the vast majority of newly diagnosed patients are expected to respond to initial treatments that incorporate novel targeted agents such as inhibitors of B cell signalling pathways, these strategies are rarely curative.3 Further, despite these advances, and even in individuals who achieve deep molecular remissions, many individuals relapse with disease that becomes progressively more refractory to successive lines of therapy.4,5 Prolonged treatment also has significant medical, social, and economic costs, and patients who become resistant have a very poor prognosis.
Cellular immunotherapies have emerged as exciting and effective therapeutic options for patients. CAR T cell therapy in particular has demonstrated remarkable clinical efficacy, leading to sustained remissions in a large percentage of treated patients with relapsed and refractory leukaemia and lymphoma.6-8 With these therapies, a patient’s T cells are typically engineered ex vivo to express a chimeric receptor containing an extracellular single-chain variable fragment fused to intracellular signalling domains, comprised of CD3ζ and costimulatory domains (e.g., CD28 or 4-1BB).9 The single-chain variable fragment portion of the synthetic receptor binds antigens on the surface of tumour cells, initiating signalling through CD3ζ, enhanced by costimulatory signalling, to elicit T cell mediated cytotoxicity. The successes of several CAR T cell clinical trials have paved the way for five U.S. Food and Drug Administration (FDA)-approved CAR T cell therapies, three of which have also gained European Medicines Agency (EMA) approval. Four of these therapies (Kymriah [tisagenlecleucel], Yescarta [axicabtagene ciloleucel], Tecartus [brexucabtagene autoleucel], and Breyanzi [lisocabtagene maraleucel]) target CD19, a B cell lineage marker present on malignant and healthy B cells.10 These therapies are approved for treatment of relapsed/refractory B cell acute lymphoblastic leukaemia (B-ALL), diffuse large B cell lymphoma, mantle cell lymphoma, and/or large B cell lymphomas.11-14 Recently, Abecma (idecabtagene vicleucel) was also approved by the FDA as the first CAR T cell therapy to target B cell maturation antigen for patients with multiple myeloma who progress or do not respond to at least four prior lines of therapy.15
Despite the great success of CAR T cell therapies for B cell malignancies, much still needs to be done to generalise this approach for the treatment of various other haematologic cancers. The sub-optimal quality of starting autologous T cells across different patients, long manufacturing processes that inadvertently exclude individuals with rapid disease progression, high production costs, and logistical issues associated with manufacturing a living, self-replicating therapeutic agent represent major hurdles to the broader translation of this approach. Additional challenges that must be overcome include exhaustion and premature senescence of CAR T cells that occur post-infusion and often result in poor proliferation, persistence, and effector function. In the setting of research efforts to enhance the quality and availability of CAR T cell therapies that are currently only available as ‘bespoke’ products, precision gene editing has emerged to produce a new generation of CAR T cells that overcome many of the aforementioned challenges. Possibilities include genetic editing strategies to improve CAR T cell anti-tumour activity and proliferative capacity, generation of allogeneic CAR T cells to circumvent autologous T cell quality challenges and enable on-demand treatment for patients, and the potential to overcome on-target, off-tumour toxicity in the context of non-B-cell haematopoietic malignancies (Figure 1).
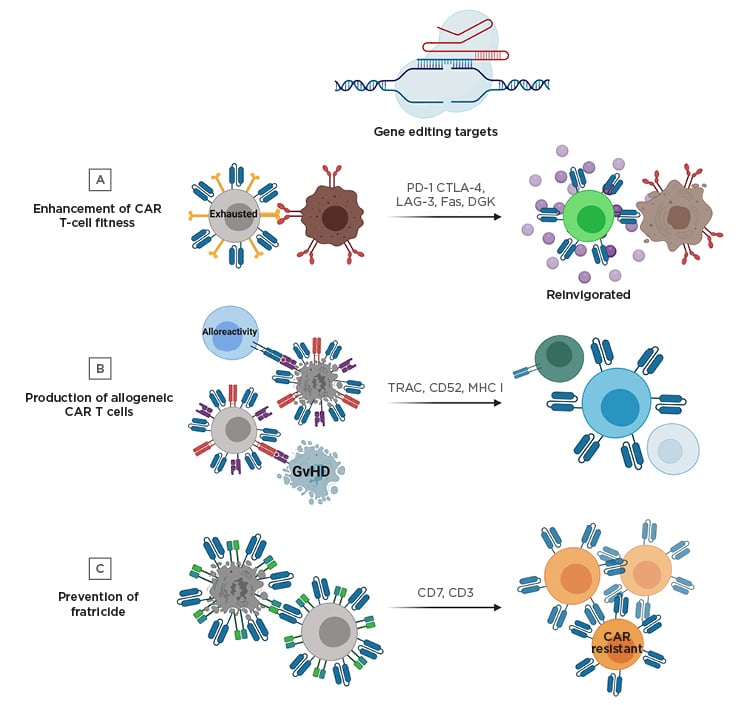
Figure 1: Gene editing strategies to improve chimeric antigen receptor T cell therapy.
A) Targeted genetic knockout of negative regulatory molecules (e.g., PD-1) can prevent exhaustion in CAR T cells and improve effector function. B) Unmatched donor-derived CAR T cells have the potential to cause GvHD and alloreactivity. To manufacture allogeneic CAR T cells that will not cause GvHD and can avoid eradication by recipient immune cells, the TCR and additional molecules can be knocked out. C) Engineering CAR T cell therapies targeting T cell lineage antigens can result in fratricide during the manufacturing process; however, this can be prevented by gene editing of the targeted antigen (e.g., CD7 or CD3).
Created with BioRender.com
CAR: chimeric antigen receptor; CTLA-4: cytotoxic T-lymphocyte-associated protein 4; DGK: diacylglycerol kinase; GvHD: graft-versus-host disease; LAG-3: lymphocyte-activation gene 3; MHC I: major histocompatibility complex Class I; PD-1: programmed cell death protein 1; TCR: T cell receptor; TRAC: T cell receptor α constant.
Gene editing has come of age over the past few decades through the development of a variety of site-specific programmable nucleases. These technologies (including zinc finger nucleases [ZFN], transcription activator-like effector nucleases [TALEN], and clustered regularly interspaced short palindromic repeats [CRISPR]-Cas9) create double-stranded breaks in the genome that can be repaired by the cell through two pathways, non-homologous end-joining (NHEJ) or homology-directed repair (HDR) (Figure 2). NHEJ is an error-prone repair pathway by which the non-homologous broken ends are ligated together, often resulting in insertions or deletions that can prevent expression of a functional protein or protein ablation. HDR occurs less frequently than NHEJ but allows for template-directed repair to facilitate site-specific insertion of a genetic sequence of choice. Although each gene editing tool has been used for both NHEJ and HDR-mediated editing, these modalities have different features with distinct advantages and limitations, as discussed below.
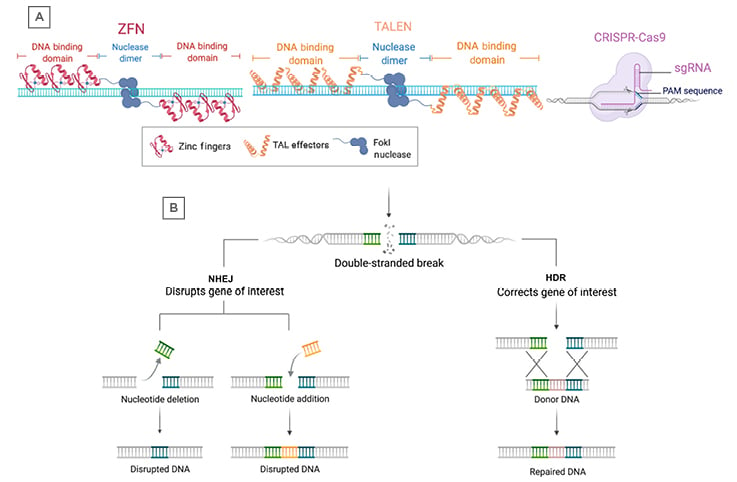
Figure 2: Programmable nucleases and DNA repair pathways.
A) The three major gene editing technologies include ZFNs, TALENs, and CRISPR-Cas9. Each programmable nuclease recognises DNA sequences through different mechanisms, including zinc finger binding domains (ZFNs), TAL effector binding domains (TALENs), or via a single guide RNA (CRISPR-Cas9). ZFNs and TALENs rely on FokI nucleases fused to the DNA binding domains that dimerise to produce double-strand breaks. The CRISPR-Cas9 system utilises the Cas9 protein, which can produce double-strand breaks without dimerisation. B) Following a double-stranded break due to a programmable nuclease, the DNA is repaired through either NHEJ or HDR. NHEJ can result in deletion or insertion of nucleotides around the break-site, leading to disruption of the target gene. HDR can use donor DNA for template-directed repair and results in insertion of a sequence of interest at the break-site.
Created with BioRender.com.
CRISPR: clustered regularly interspaced short palindromic repeats; HDR: homology-directed repair; NHEJ: non-homologuous end joining; TALENS: TAL: transcription activator-like effector binding domains; ZFN: zinc finger nuclease.
A ZFN is a restriction enzyme fused to zinc finger motifs that can recognise a triplet codon sequence in DNA. Although ZFNs can have good editing efficiency, they are often difficult to engineer and exploit for multiplex gene editing and are not highly specific.16 TALENs (consisting of TAL proteins fused to a nuclease) are much easier to engineer, have higher editing efficiency compared to ZFNs, and display very low off-target editing. However, they are difficult to deliver to cells and have limited high throughput editing capacity.16 Finally, the CRISPR-Cas9 system consists of an RNA-guided endonuclease (Cas9), which is directed to DNA cut-sites with a high degree of specificity and efficiency. As the Cas9 protein is directed by a single guide RNA (sgRNA), this system is simple to employ and can easily be used for editing multiple genes; however, conventional CRISPR-Cas9 systems tend to exhibit higher off-target editing rates compared with TALENs.16
STRATEGIES TO IMPROVE CHIMERIC ANTIGEN RECEPTOR T CELL FITNESS
Inhibitory receptors such as programmed cell death protein 1 (PD-1), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), lymphocyte-activation gene 3 (LAG-3), and T cell immunoglobulin and mucin domain 3 are natural regulators that limit T cell activation and prevent autoimmunity as well as severe inflammation. These molecules have also been demonstrated to hamper anti-tumour immune responses and potentiate tumour escape through binding to their cognate ligands overexpressed on malignant cells.17 Antibodies employed to interfere with inhibitory receptor activation have proven effective for therapy of both liquid and solid tumours.18 These therapies can act synergistically with CAR T cell therapies by preventing or reversing CAR T cell inhibition.18 However, antibody-based checkpoint blockade therapies often require repeated administration to maximise efficacy and have been associated with severe toxicities, particularly autoimmune reactions.19
Genetic ablation of inhibitory molecules in CAR T cells is an alternative strategy that would avoid the systemic toxicities of conventional immune checkpoint blockade approaches. For example, knockout of PDCD1 (gene coding for PD-1) has been widely employed for this purpose and the strategy has frequently augmented CAR T cell proliferation, cytotoxicity, and overall anti-tumour activity in multiple preclinical models of both haematopoietic and non-haematopoietic cancers.20-23 It is important to note that these enhancements to antitumour efficacy are dependent on the expression of programmed death-ligand 1 (PD-L1) on tumour cells and most of these model systems require forced expression of PD-L1; however, Ren et al. found that over 90% of naïve prostate tumour (PC3) cells gain PD-L1 expression after encountering prostate stem cell antigen-directed CAR T cells, suggesting that PD-1 ablation may help avoid adaptive resistance resulting from upregulation of inhibitory ligands on tumour cells.20 Additionally, PD-1 knockout appears to only restore anti-tumour function impaired by PD-1/PD-L1 interactions back to baseline levels, but does not enhance cytotoxic CAR T cell capacity against PD-L1 negative cell lines. There are also conflicting reports regarding the sustained T cell potency enhancing effects associated with PD-1 genetic ablation, with one study reporting a significant increase in terminally differentiated CD8+ T cells and a long-term reduction in survival, proliferation, and durability of PD-1 knockout cells in a model of chronic virus infection.24 Dissecting the effects of PD-1 ablation is difficult due to its complex role in both the negative regulation of T cell function as well as the maintenance of T cell effector activity, persistence, and memory differentiation.25 Further investigation is required to determine if the benefit of short-term improvement in CAR T cell proliferation and anti-tumour function outweighs the potential for inducing T cell hypofunction due to PD-1 disruption. Despite these conflicting reports, many cellular therapy clinical trials are currently being conducted to include CAR T cell-intrinsic PD-1 inhibition strategies.
In addition to PD-1, disruption of other negative regulators of CAR T cell function, including LAG-3, CD95/Fas, and diacylglycerol kinase (DGK), have been investigated. Unexpectedly, genetic deletion of the LAG-3 inhibitory receptor in anti-CD19 CAR T cells does not improve antitumor efficacy.26 However, CAR T cell-intrinsic CD95/Fas ablation has demonstrated some success. Although T cells utilise Fas signalling as a mechanism to induce apoptosis of cancerous and infected cell targets, this death pathway may also inhibit anti-tumour T cell activity.27,28 Accordingly, blockade of Fas signalling restores anti-tumour T cell function, and CD95/Fas genetic ablation leads to reduced apoptosis and increased expansion of anti-CD19 CAR T cells.27,29 Finally, ablation of DGK enhances CAR T effector functions, resulting in resistance against immunosuppressive factors, and increased durability of cytotoxic functions following repeated stimulation in models of glioblastoma and mesothelioma.30,31 This was somewhat expected, as DGK is known to restrict T cell activation by the metabolisation of the second messenger, diacylglycerol, resulting in negative regulation of antigen receptor signalling.32
Ablation of multiple inhibitory pathways has the potential to provide CAR T cells with even more enhanced durability and potency. In this regard, the feasibility of multiplex editing has been demonstrated in preclinical studies.20,29,33 A study by Ren et al. accomplished quadruple multiplex editing of CAR T cells, ablating TRAC, B2M, PDCD1, and CTLA-4 simultaneously.29 However, there are many safety considerations that must be taken into account when considering clinical translation of such a strategy. Notably, the consequences of genetic instability and translocations induced by multiple double-strand DNA breaks are not fully understood, particularly when introducing multiplex-edited cells in patients. In a Phase I clinical trial recently conducted by the authors, triple-knockout TRAC, TRBC, and PDCD1 T cell receptor (TCR)-engineered T cells targeting cancer/testis antigen 1 was a safe and feasible therapy in patients.34 This study demonstrated that although chromosomal translocations were observed during cell manufacturing, translocation frequency decreased after infusion. These translocations resulting from multiplex CRISPR-Cas9 editing were not associated with clonal expansion or abnormal proliferation of cancer/testis antigen 1 T cells, at least within 9 months following administration. However, additional clinical investigations with longer patient follow-up times need to be conducted to fully evaluate any potential safety risks.
In addition to ablation of negative regulators, genetic editing strategies can be used to integrate a variety of gene cassettes into a locus/loci of choice.35 This allows for controlled integration of a transgene, limiting the potential risk of oncogenic transformation due to random insertional mutagenesis by a viral vector. Additionally, placing a transgene under control of an endogenous promoter may allow for more regulated expression of the transgene. For example, studies have investigated integrating CAR transgenes into the TRAC locus, leading to increased CAR expression and enhanced anti-tumour potency of the engineered cells.36,37 In addition, Sachdeva et al. explored integration of proinflammatory IL-12P70 into the CD25 or PDCD1 locus, permitting regulated cytokine expression upon T cell activation. This strategy allows for tightly controlled expression of IL-12P70 following tumour cell recognition, thus augmenting anti-tumour activity through the combined proinflammatory effects of the cytokine and amelioration of PD-1-mediated negative regulation.37 This approach can be generalised to knocking cassettes encoding a wide array of effector molecules into various target loci, opening up opportunities to direct location-specific anti-tumour mediator expression to potentiate CAR T cell therapy.
A variety of other potential gene editing targets to improve CAR T cell function are currently being explored, including modulation of proteins involved in T cell fate determination. The authors found that accidental ablation of tet methylcytosine dioxygenase 2 (TET2), a protein involved in DNA demethylation, skews CAR T cell differentiation toward a central memory state and confers enhanced proliferation and anti-tumour function.38 In this ‘bedside-back-to-bench’ study, TET2 disruption altered global and site-specific chromatin accessibility and transcription of genes involved in cell cycle progression and T cell receptor signalling. The resultant clonally expanded CAR T cells possessed properties of short-lived memory cells with potent effector functions and long-lived memory cells with long-term persistence. Despite this remarkable effect, TET2 may not be a viable target for conventional gene editing due to its well-characterised function as a tumour suppressor. Future studies will evaluate the potential benefit of targeting alternative genes involved in regulation of T cell differentiation, effector function, and persistence to enhance the effectiveness of CAR T cell therapies.
THE PROMISE OF UNIVERSAL CHIMERIC ANTIGEN RECEPTOR T CELLS
The generation of ‘off-the-shelf’ allogeneic CAR T cell products has been a major goal in the cellular immunotherapy field. Engineering CAR T cells from healthy donors would solve many logistical issues, allowing for rapid treatment of patients and major reductions in manufacturing costs. Additionally, because these T cells would be derived from healthy donors, they may not be dysfunctional at the start of the manufacturing process, unlike the case with many conventional autologous cell therapy approaches. However, there are several challenges with developing this kind of therapy. To achieve safe and effective treatment, universal CAR T cells must not induce graft-versus-host-disease (GvHD) and must be resistant to host-versus-graft alloreactivity. Many strategies have been investigated to accomplish these goals using gene editing technologies.
Elimination of GvHD has primarily been achieved through genetic disruption of endogenous TCR expression in CAR T cells. In the first proof-of-concept study of the feasibility of engineering allogeneic CAR T cells ZFNs were used to knockout the TCRα- or β-chain (i.e., through TRAC or TRBC targeting). This work revealed that TCR knockout does not significantly impact the cytotoxic functions or proliferative capacities of edited CAR T cells.39 The study thus opened the door for development of allogeneic CAR T cells, with TRAC and/or TRBC genetic disruption being used in a multitude of preclinical and clinical investigations.20,33,34,40-43,44 Investigators have also pursued approaches to permit site-specific integration of CARs into the TRAC locus to safely insert the synthetic receptor transgene while simultaneously eliminating GvHD, improving CAR expression and enhancing CAR T cell potency.36,41
In addition to prevention of GvHD, many studies have also investigated strategies to prevent endogenous recipient immune cells from eliminating allogeneic CAR T cells. One method has been to engineer CAR T cells that are resistant to lymphodepletion. TALEN-editing of TRAC and CD52 leads to generation of CAR T cells that do not cause GvHD and are rendered resistant to alemtuzumab-mediated lymphodepletion targeted against CD52+ wild-type alloreactive T cells. This strategy has been evaluated preclinically in the setting of B cell maturation antigen-directed CAR T cells for multiple myeloma.40 In addition, this strategy has been used for the development of universal CD19-targeting CAR T cells (UCART19). UCART19 has demonstrated powerful clinical efficacy, achieving molecular remission in two paediatric patients with B-ALL.43 Following the success observed in paediatric patients, UCART19 was then expanded to two multicentre Phase I clinical trials evaluating feasibility, safety, and efficacy in adult and paediatric patients with B-ALL (NCT02808442,45 NCT0274695246). These trials showed cytokine release syndrome (CRS) as the most common adverse event (91% of patients; 14% had Grade 3–4 CRS) and limited GvHD (10% of patients), demonstrating a manageable safety profile.44 Fourteen of the 21 patients had a complete response or complete response with incomplete haematological recovery at 28 days post-UCART19 infusion. Progression-free survival was 27% at 6 months, demonstrating initial clinical efficacy of UCART19 cells in aggressive B cell leukaemia.
An additional strategy to avoid host destruction of allogeneic CAR T cells is elimination of human leukocyte antigen Class I expression to prevent recognition of CAR T cells by recipient T cells. Disruption of human leukocyte antigen-A has been shown to allow for allogeneic CAR T cell escape from host T cell-mediated killing.47 Other studies have targeted B2M, a component of major histocompatibility complex Class I molecules. Multiplexing B2M knockout with TRBC and PDCD1 disruption leads to CAR T cells with reduced alloreactivity, elimination of GvHD, and enhanced in vivo anti-tumour activity.20 Multiplex editing combining knockout of the endogenous TCR together with a molecule to prevent alloreactivity (e.g., CD52 or B2M) will be required to manufacture an off-the-shelf allogeneic CAR T cell product, and inclusion of additional genetic edits are likely needed to enhance clinical efficacy.
CHIMERIC ANTIGEN RECEPTOR T CELL THERAPY FOR NON-B CELL MALIGNANCIES
Antigen-directed treatment of haematologic malignancies is challenging due to the sparsity of conserved tumour-specific targets. CAR T cell therapies for haematologic malignancies typically target cell lineage markers, such as CD19 for B cell malignancies. Thus, CAR T cell therapy for these neoplasms results in destruction of both malignant and healthy cells, which can result in severe toxicity. Targeting B cell malignancies has had greater success compared to other haematologic neoplasms due to well-tolerated and effectively managed B cell aplasia.48 However, the targeting of other haematologic malignancies such as myeloid and T cell leukaemias can result in lethal toxicities due to on-target, off-tumour effects. Current gene editing research in the context of cellular therapies has, therefore, been directed toward developing strategies to avoid these limitations.
Myelotoxicity is a prominent concern when translating CAR T cell therapies to treat myeloid malignancies. One strategy to avoid CAR T cell-mediated destruction of healthy myeloid cells is to knockout the targeted lineage antigen in haematopoietic stem cells (HSC) and transplant these cells into patients after CAR T cell therapy. This allows for repopulation of the myeloid compartment with CAR-resistant cells. Studies have investigated this approach for anti-CD33 CAR T cells and found that genetic ablation of CD33 in haematopoietic stem and progenitor cells can be used for HSC transplantation. This strategy led to generation of a CAR-resistant myeloid system that allows for selective targeting of cancer cells without myelotoxicity.48,49 The method could be translated to other therapies targeting myeloid malignancies such as CD123-directed CAR T cells. CD123 is overexpressed on a variety of haematologic malignancies, and CD123 CAR T cells are being investigated for treatment of myelodysplastic syndrome, blastic plasmacytoid dendritic neoplasm, and acute myeloid leukaemia, with a clinical trial currently underway for relapsed/refractory acute myeloid leukaemia (NCT0319027850).51,52 Implementation of subsequent HSC transplantation with CD123 deficient cells could prevent toxicities associated with destruction of healthy CD123+ myeloid cells and offers a feasible treatment for myeloid malignancies.
CAR T cell therapies for T cell malignancies have proven difficult to translate clinically due to targeting of T cell lineage antigens shared by both normal and malignant cells, which are also expressed on CAR T cells. This leads to ‘fratricide’ during the CAR T cell manufacturing process, in which CAR T cells induce cytotoxicity of other engineered T cells. One strategy for fratricide-resistant CAR T cell generation is genetic depletion of CD7 in CD7-targeting cells. Preclinical studies have demonstrated anti-tumour efficacy of allogeneic CAR T cells with CD7 and TRAC knockout.53 The use of an allogeneic system reduces the risk of generating CD7-negative, CAR-resistant leukaemic T cells, which is a major safety concern with an autologous product. Initial clinical evaluation of an allogeneic CD7-targeting CAR T cell product has demonstrated safety and efficacy in two patients with T cell acute lymphoblastic leukaemia (T-ALL).54 Accordingly, a Phase I clinical trial is being initiated with autologous CRISPR-edited, CD7-targeting CAR T cells for T-ALL and T cell non-Hodgkin lymphoma (NCT0369001155). In addition to CD7-targeting CAR T cells, CD3-targeting CAR T cells have been investigated for certain T cell malignancies. A study showed that ablation of CD3 and TRAC led to the generation of CD3e-CAR resistant T cells, allowing for the production of CD3e-targeting CAR T cells for T cell leukaemias.56 Although this system would solve manufacturing issues, more research is needed to investigate the feasibility and management of potential T cell compartment depletion in patients with T-ALL with such an approach.
CHALLENGES AND EXPANDING OPPORTUNITIES WITH GENOME EDITING OF CHIMERIC ANTIGEN RECEPTOR T CELLS
Despite the promise that gene editing has shown in both preclinical and clinical studies, there are many challenges and concerns that need to be addressed to ensure the safety and reliability of this strategy. Some of these obstacles include severe clinical adverse events due to disruption of negative regulatory pathways, low knockout efficiencies, genetic instability caused by double strand DNA breaks, and the risk of off-target editing.
Major adverse events have been observed in the context of CAR T cell therapies, including neurotoxicity and CRS. Severe CRS can be fatal and is characterised by a massive production of proinflammatory cytokines, particularly IL-6.57 Inhibitors of IL-6 are used for the management of severe CRS during CAR T cell treatment; however, there is concern that administration of these inhibitors could negatively impact the anti-tumour activity of CAR T cells.57 When developing edited CAR T cells that disrupt negative regulatory pathways such as PD-1 there is concern for potential development of more severe CRS. As the understanding of the pathophysiology of CRS continues to expand, new genetic editing strategies have emerged to prevent or diminish CRS. For example, neutralisation of granulocyte-macrophage-colony-stimulating factor (GM-CSF) leads to a reduction in toxicities associated with CAR T cell therapy, particularly neuroinflammation and CRS.58 One study found that genetic ablation of GM-CSF in CAR T cells results in a drastic decrease in production of a variety of several proinflammatory cytokines, without impacting the anti-tumour function or proliferation of the engineered cells in vitro.59 An additional study found that GM-CSF-deficient CD19 CAR T cells have enhanced anti-tumour potency, leading to improved survival in xenogeneic mouse models.58
To address issues of poor editing efficiency, improved CAR transgene delivery methods and editing platform advancements have been under investigation. Traditional viral transduction of CAR T cells followed by introduction of CRISPR-Cas9 editing machinery typically produces a heterogeneous cell population, in which not all CAR T cells are edited. This poses a problem for universal CAR T cell approaches, as knockout of the endogenous TCR is necessary to prevent GvHD, as discussed above. To improve the generation of universal CAR T cells, Georgiadis et al. created a Terminal-TRAC CAR T system in which a self-inactivating lentiviral vector delivered the CAR transgene and sgRNA targeting TRAC, coupling CAR integration with CRISPR-Cas9 editing.42 This ensures that all T cells transduced with the CAR are TRAC deficient, eliminating safety concerns associated with incomplete TCR knockout in the CAR+ population. In addition to employing plasmid-delivery systems to eliminate safety concerns of incomplete editing in universal CAR T cells, improved plasmid delivery methods have been used to develop simplified engineering processes and achieve more efficient genome editing. Hu et al. utilised a platform of nucleofection to introduce plasmids encoding a CD133-targeted CAR and CRISPR-Cas9 machinery for PDCD1 knockout to achieve CAR integration and gene editing in a single reaction.23 This simplified strategy of simultaneously delivering the CAR transgene and CRISPR-Cas9 editing machinery provides an efficient process for engineering cells that would be more desirable for manufacturing compared to the current multi-step process. Plasmid-based delivery can also enhance gene editing efficiency. A limiting factor of editing efficiency by CRISPR is fast degradation of sgRNAs compared to Cas9 protein or mRNA, and higher editing can be achieved by constitutive expression of the sgRNAs. By using a CAR lentiviral vector incorporating TRAC-sgRNA, Ren et al were able to achieve high percentages of CD3-disrupted cells.29 This system can be used for multiplex editing as well, with triple- and quadruple-knockouts targeting TRAC, B2M, Fas, PDCD1, and/or CTLA-4.29
Editing technologies such as CRISPR-Cas9, TALENs, and ZFNs achieve gene knockouts through double-stranded breaks in the DNA. These breaks are mended through an error-prone DNA repair pathway, resulting in insertions and deletions which ideally prevent expression of functional protein. This process allows little control over how the cell is edited and has an increased risk of off-target editing. As such, base editing has emerged as a potentially more precise alternative to classic CRISPR-Cas9 editing. Base editors consist of a catalytically inactive Cas9 nuclease fused to DNA deaminase that can introduce site-specific point mutations through conversion of nucleotide bases without double-stranded breaks.60 This allows for enhanced control over genetic disruption through altering bases to introduce premature stop codons or affect splice sites.61-63 Additionally, because base editing does not result in double stranded breaks in the genome, it is a safer option to prevent translocations associated with multiplex editing. Webber et al. utilised base editing to create allogeneic CAR T cells by introducing premature stop codons or impacting splice sites in PDCD1, TRAC, and B2M.64 This study revealed that multiplex base editing in primary human T cells resulted in reduced double stranded breaks and undetectable translocations without altering CAR T cell function.
In addition to chromosomal translocations, off-target editing is a major safety concern when translating CRISPR-Cas9 technology into the clinic. There are numerous methods to evaluate off-target editing, including homology-dependent and homology-independent approaches, which are often used in combination. Homology-dependent methods involve computational strategies that evaluate potential off-target sequences based on presence of a protospacer adjacent motif sites and similarity to the sgRNA sequence. Homology-independent methods are empirical measurements of off-target editing throughout the whole genome that employ a variety of assays coupled with next-generation sequencing. Some homology-independent approaches are genome-wide unbiased identification of double-stranded breaks enabled by sequencing (GUIDE-seq), discovery of in situ Cas off-targets and verification by sequencing (DISCOVER-seq), circularisation for in vitro reporting of cleavage effects by sequencing (CIRCLE-seq), cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq), and Digenome-seq.65-69 The challenge with using these assays is obtaining genome-wide coverage with enough sensitivity to detect rare off-target events and avoiding biases in the evaluation of off-target editing. Each of these methods possesses unique strengths and weaknesses and can be used in parallel to evaluate off-target editing more deeply. A recent comparison of three homology-independent methods (GUIDE-seq, CIRCLE-seq, and CITE-seq) showed similar accuracy in the detection of off-target CRISPR-Cas9-mediated gene editing.70 The authors gave guidance in assessing off-target effects and suggested that GUIDE-seq may be the more appropriate tool to evaluate ex vivo engineered cells, such as CAR T cells. Additionally, novel techniques for evaluating off-target effects are actively being developed and optimised such as amplification-free long-read sequencing.71 Thus, clinical translation of gene-edited cellular therapeutics will likely incorporate a variety of strategies to accurately detect off-target mutations and thus allay associated safety concerns.
CONCLUDING REMARKS
Despite the clinical successes of CAR T cell therapy in the setting of certain haematological malignancies, further development of this approach is necessary to achieve similar efficacy in other cancers. Gene editing has the potential to greatly improve CAR T cell therapies by enhancing T cell fitness, enabling the manufacture of off-the-shelf products, and allowing for treatment of T cell and myeloid cell cancers. Advances in multiplex gene editing are particularly exciting, as several modifications will allow for development of robust allogeneic CAR T cell treatments for many different diseases. However, safety concerns must be addressed to effectively integrate single and multiplex gene editing into the production of cellular therapeutics. The advent of more precise gene editing technologies such as base editing has the potential to produce safe and efficient multiplex editing strategies for this purpose. These innovations enable greater control over genetic modifications to CAR T cells, addressing many potential safety issues associated with genomic instability and off-target editing. Developments in the next few years in the areas of feasibility, safety, and efficacy of gene-edited CAR T cell products will ultimately determine how far reaching this approach will be in the broader battle against cancer.





