
Abstract
Improved Clinical, Laboratory, Molecular, and Pathological (CLMP) 2017 criteria for myeloproliferative neoplasms (MPN) define the JAK2V617F trilinear MPNs as a broad continuum of essential thrombocythaemia (ET), polycythaemia vera (PV), masked PV, and post-ET or post-PV myelofibrosis (MF). Normal versus increased erythrocyte counts (5.8×1012/L) on top of bone marrow histology separate JAK2V617F ET and prodromal PV from early and classical PV. Bone marrow histology of the JAK2V617F trilinear MPNs show variable degrees of normocellular megakaryocytic, erythrocytic megakaryocytic and erythrocytic megakaryocytic granulocytic (EMG) myeloproliferation, peripheral cytoses, and splenomegaly related to JAK2V617F allele burden. MPL515 thrombocythaemia displays predominantly normocellular megakaryocytic proliferation. CALR thrombocythaemia intially presents with megakaryocytic followed by dual granulocytic and megakaryocytic myeloproliferation without features of PV. The megakaryocytes are large, mature, and pleomorphic with hyperlobulated nuclei in JAK2V617F ET and prodromal, classical, and masked PV. The megakaryocytes are large to giant with hyperlobulated staghorn-like nuclei in MPL515 thrombocythaemia. The megakaryocytes are densely clustered, large, and immature dysmorphic with bulky (bulbous) hyperchromatic nuclei in CALR thrombocythaemia and MF.
INTRODUCTION
The diagnostic clinical and bone marrow criteria of polycythaemia vera (PV) between 1940 and 1950 were plethoric appearance, splenomegaly, elevated erythrocyte count >6×1012/L, elevated platelet count, elevated haematocrit (Ht), and pathognomonic bone marrow features showing a panmyelosis with increased erythrocytic megakaryocytic granulocytic (EMG) trilinear haematopoiesis.1 About one-third of PV patients develop splenomegaly and myelofibrosis (MF) after follow-up of 15–30 years.2,3 The combination of a persistent increase of platelet counts (>350×109/L) and a monolinear proliferation of large mature megakaryocytes in the bone marrow is diagnostic for essential thrombocythaemia (ET).4 Dameshek3 speculated on the possible causal interrelation among myeloproliferative disorders (MPD) showing trilinear bone marrow features in PV, the dual increase of megakaryopoiesis in agnogenic myeloid metaplasia (AMM), and unilinear megakaryopoiesis in megakaryocytic leukaemia (ML) without PV features in the bone marrow (Figure 1).3 The Polycythemia Vera Study Group (PVSG) criteria for the clinical diagnosis of PV do not include a bone marrow biopsy to differentiate between PV and erythrocytosis and overlook idiopathic erythrocythaemia by definition.5,6 Idiopathic erythrocythaemia is distinguished by increased red cell mass (RCM), normal platelet count and leukocyte count, and no splenomegaly on palpation.5,6 Lamy et al.7 measured RCM in 85 patients meeting the PVSG criteria for PV (haemoglobin [Hb] >18 g/dL, Ht >0.52 in males and Hb >16 g/dL, Ht >0.47 in females) and in 18 patients with masked PV (inapparent PV) with normal erythrocyte, Hb, and Ht values. RCM was increased in patients with classical PV with no or minor splenomegaly. RCM was increased in masked or inapparent PV patients due to significant splenomegaly and hypersplenism.7 The PVSG defined idiopathic or AMM of spleen and bone marrow as primary MF (PMF). As MF is a secondary event in all variants of MPD, the Hannover and Rotterdam Bone Marrow Classification discovered prefibrotic ML and fibrotic stages of AMM as the third distinct MPD entity of primary megakaryoctic granulocytic myeloproliferation (PMGM) without features of PV (Figure 1).4
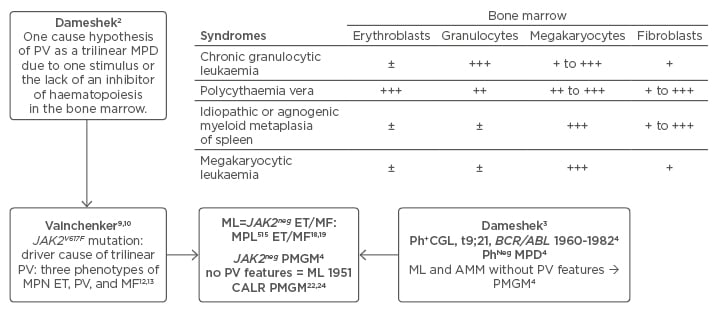
Figure 1: Changing concepts of PVSG-WHO classification of myeloproliferative neoplasms into CLMP criteria of JAK2V617F mutated essential thrombocythaemia, polycythaemia vera, and myelofibrosis and JAK2 wildtype CALR or MPL mutated thrombocythaemia and myelofibrosis: from Dameshek to Vainchenker, Kralovics and Michiels 1950–2017.
Left) The Dameshek (1950) one cause hypothesis of PV as a trilinear MPD2 has been confirmed by Vainchenker’s discovery in 2005 of the JAK2V617F mutation8 as the driver of the trilinear MPNs, ET, PV, and MF.9,10,11,33 Upper) Dameshek (1951)3 speculated on the possible causal interrelationship among MPS showing trilinear bone marrow features in PV, dual increase of megakaryocytes and fibroblasts in AMM and unilinear megakaryopoiesis in ML. Right middle) The Hannover and Rotterdam Bone Marrow Classifications of the MPD recognised prefibrotic ML and AMM as a distinct entity of prefibrotic and fibrotic stages of PMGM without features of PV.4 Bottom) ML defined by Dameshek in 1951 can readily be translated into JAK2-negative MPL mutated ET and MF and CALR ET associated with PMGM without features of PV.
ET: essential thrombocythaemia; MF: myelofibrosis; PV: polycythaemia vera; MPN: myeloproliferative neoplasms; MPD: myeloproliferative disorders; ML: megakaryocytic leukaemia; MPS: myeloproliferative syndromes; AMM: agnogenic myeloid metaplasia; PMGM: primary megakaryocytic granulocytic myeloproliferation.
MOLECULAR AETIOLOGY OF JAK2V617F TRILINEAR MYELOPROLIFERATIVE DISORDERS
The one cause hypothesis of Dameshek2 that PV is a trilinear MPD has been confirmed by Vainchenker’s discovery in 2005 that the acquired JAK2V617F mutation is the driver cause of three MPD phenotypes: ET, PV, and MF (Figure 1).8-10 Vainchenker and Constantinescu9 proposed the concept that low V617F constitutional kinase activity in heterozygous mutated JAK2V617F mutated patients is enough to produce the ET phenotype and that higher V617F constitutional kinase activity in JAK2V617F mutated heterozygous/homozygous or homozygous mutated patients is needed to produce the PV phenotype (Figure 2).9,10 The JAK2V617F dosage hypothesis has been confirmed at the bone marrow haematopoietic stem cell level by the demonstration that endogenous erythroid colonies (EEC) from ET patients are mainly heterozygous for the JAK2V617F mutation, whereas all PV patients are either hetero/homozygous or mainly homozygous for the JAK2V617F mutation (Figure 2).11
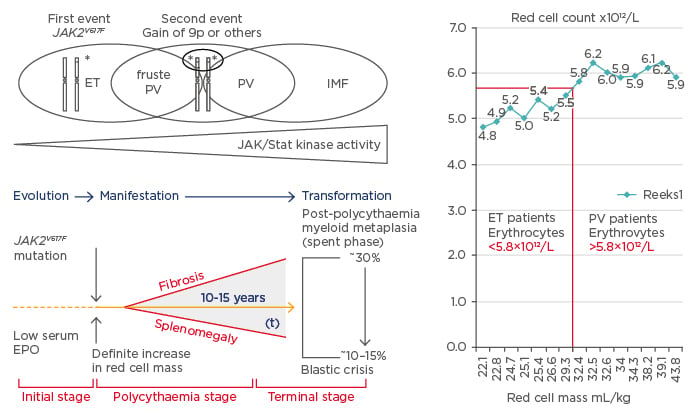
Figure 2: The sequential occurence of CLMP defined essential thrombocythaemia, polycythaemia vera, and myelofibrosis related to JAK2 allele burden in JAK2V617F mutated trilinear myeloproliferative neoplasms.
Upper) The discovery of the somatic JAK2V617F gain mutation can explain the three sequential phenotypes of ET, PV, and MF. A slight increase (changes) in the JAK2V617F kinase activity in heterozygous mutated MPN is enough to produce the clinical phenotype of ET. Increasing levels of JAK2V617F kinase activity in trilinear MPN due to mitotic recombination resulting in heterozygous/homozygous and predominantly homozygous mutated MPN is associated with early, overt, and advanced PV, respectively (Vainchenker and Constantinescu 2005,9 Villeval et al. 2006.10 Lower) Dynamics of the JAK2V617F disease processes in PV as a broad spectrum (Tables 1 and 2) ranging from normocellular ET, prodromal PV mimicking ET and the defintive increase in red cells (>5.8×1012/L) followed by masked PV, PV complicated by fibrosis and splenomegaly, spent phase PV and blastic transformation. Designed by Michiels et al. 2006–2016. Right) Initial stage of JAK2V617F mutated ET and prodromal PV with normal RCM and erythrocytes <5.7×1012/L versus manifest PV with definitive increase of RCM and erythrocytes >5.7×1012/L.4,37
EPO: erythropoietin; ET: essential thrombocythaemia; RCM: red cell mass; PV: polycythaemia vera; MF: myelofibrosis; MPN: myeloproliferative neoplasms.
Michiels and Medinger12 studied RCM in relation to erythrocyte count in World Health Organization (WHO)-defined ET patients (24 patients) and PV patients (46 patients) with no or minor splenomegaly. The JAK2V617F mutation load in 24 ET patients was zero in 10 patients and positive in 14 patients; this mutation load ranged from 3–20%, 20–42%, and >50% in six, five, and two cases, respectively. The JAK2V617F mutation load in 36 evaluable PV patients ranged from 3–20% and from 20–50% and was >50% in 5, 12, and 19 PV cases, respectively. Increased erythrocyte counts above normal levels (>5.8×1012/L in males and >5.6×1012/L in females) correlated with increased RCM in PV patients whereas ET patients had normal erythrocyte counts and RCM (Figure 2).12 Increased RCM and erythrocytes >5.8/5.6×1012/L in PV were associated with Hb values from 14.6–18.9 g/L and Ht values from 0.46–0.57. Normal RCM in ET patients were related to erythrocyte counts of 4.6–5.4×1012/L, Hb from 14.0–16.1 g/L, and Ht from 0.39–0.47,13 consistent with the diagnosis of ET or prodromal PV (Tables 1 and 2).
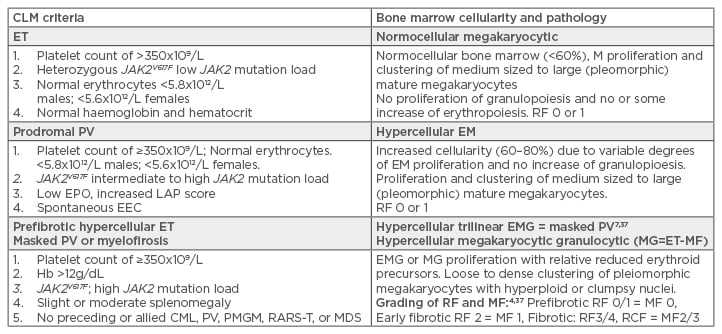
Table 1: International Clinical, Laboratory, Molecular, and Pathobiological (2017 CLMP) criteria for diagnosis of JAK2V617F mutated essential thrombocythaemia, prodromal polycythaemia vera, masked polycythaemia vera due to splenomegaly, and post essential thrombocythaemia myelofibrosis.
CLM: Clinical, Laboratory, and Molecular; ET: essential thrombocythaemia; PV: polycythaemia vera; EPO: erythropoietin; EEC: endogenous erythroid colony formation; Hb: haemoglobin; CML: chronic myeloid leukaemia; PMGM: primary megakaryocytic granulocytic myeloproliferation; MDS: myelodysplastic syndrome; RARS-T: refractory anaemia with ringed sideroblasts associated with marked thrombocytosis; EMG: erythrocytic megakaryocytic granulocytic; RF: reticuline fibrosis; MF: myelofibrosis.

Table 2: International Clinical Molecular and Pathological criteria for the diagnosis of JAK2 mutated classical polycythaemia vera, masked polycythaemia vera due to splenomegaly or exon 12 PV versus primary or secondary erythrocytoses.
CLM: Clinical, Laboratory, and Molecular; PV: polycythaemia vera; Hb: haemoglobin; Ht: haematocrit; RCM: red cell mass; EMG: erthrocytic megakaryocytic granulocytic; RF: grading fibrosis; MF: myelofibrosis.
The mutation load in percentages of JAK2 mutated granulocytes in a large retrospective Italian study of JAK2V617F trilinear MPNs was low in 250 ET patients (median: 18%), significantly higher in 212 PV patients (median: 42%) and 18 post-ET MF patients (median: 42%), and predominantly high (>50%) in post-PV MF (median: 93%) patients.14 A JAK2 allele burden >50% (homozygous) was recorded in 2% of 250 ET patients, in 41% of 212 PV patients, in 72% of 18 post-ET patients, and in 93% of 55 post-PV patients.14 The correctness of the JAK2 dosage hypothesis has been confirmed in patients with hereditary ET caused by the heterozygous germline gain of function mutations JAK2V617I and JAK2R564Q in the JAK2 gene.15-17 Affected hereditary ET patients heterozygous for the JAK2V617I and JAK2R564Q germline mutations have a clinical ET phenotype with normal values for Hb, Ht, erythrocytes, thrombopoietine (TPO), and erythropoietin (EPO) levels. The response to EPO in the EEC assay was normal in congenital JAKV617I and JAK2R564Q but increased in acquired JAK2V617F.15-17
JAK2 WILDTYPE MPL515 MUTATED MEGAKARYOCYTIC LEUKAEMIA OR ESSENTIAL THROMBOCYTHAEMIA (FIGURE 1 )
With the advent of the JAK2V617F discovery, two variants of JAK2neg MPN have been discovered: MPL515 mutated ET and MF4,18,19 and CALR mutated ET and MF in PMGM patients.19-23 (Figure 1). MPLW515L and MPLW515K as the driving cause of MPN in large series of ML, or ET and MF patients occurred with a frequency rate of approximately 1% and 5%, respectively.18,19 In a European study19 of 176 cases with the MPL515 mutation, the MPLW515L mutation occurred in 110 cases, and the MPLW515K mutation in 58 cases. The overall mutation levels were lower (25%) in MPLW515L (n=106) compared with the level of 37% in cases with MPLW515K (n=32). Of the 138 MPL515 cases (ET, n=99; MF, n=36; ratio of ET versus MF: 2:1), the median MPLW515L mutation levels were significantly lower (21%) in ET than those (46%) in MF patients. The 29 homozygous MPL515 positive cases had a diagnosis of MF in 15 patients and ET in 12 patients.
The presence of clustered small and giant megakaryocytes with deeply lobulated staghorn like nuclei in MPL515 mutated ET are not seen in JAK2V617F positive normocellular ET, prodromal PV, masked PV, and PV.20,21 The pleomorphic megakaryocytes in JAK2V617F mutated ET in bone marrow biopsy were not larger but similar in size to medium to large megakaryocytes (pleomorphic) in prodromal and overt PV. Erythropoiesis in MPL thrombocythaemia is reduced whereas a local increase of erythropoiesis in areas of loose clustered pleiomorphic megakaryoctyes is present in JAK2V617F normocelluar ET and prodromal PV. LAF score, serum EPO, and ferritin levels are normal in MPL MPN cases and increased in JAK2V617F MPN cases.20,21
MEGAKARYOCYTIC LEUKAEMIA AND CALR THROMBOCYTHAEMIA WITHOUT POLYCYTHAEMIA FEATURES
CALR as the driving cause of ML3 or PMGM4 (Figure 1) has been detected in the majority of JAK2neg WHO-defined ET and PMF cases by Kralovics;22 this was the second groundbreaking event in MPN molecular research that prompted us to revise and simplify the 2016 WHO and European Clinical, Molecular and Pathological (ECMP) MPN classifications4,13,20,21 into a new set of Clinical Laboratory, Molecular and Pathologic (CLMP) criteria for JAK2, MPL, and CALR mutated MPNs (Tables 1, 2, and 3). The MPN research laboratory of Kralovics4 discovered somatic mutations of 52-bp deletion in one patient, of 1-bp deletion in one patient, and recurrent 5-bp insertion in four PMF patients.22
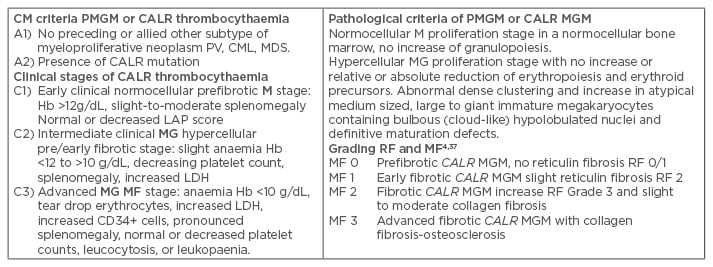
Table 3: Clinical Laboratory, Molecular and Pathological criteria for hypercellular essential thrombocythaemia associated with primary megakaryocytic, granulocytic myeloproliferation caused by calreticulin mutations.
The combination of A1 + A2 and P1 establishes JAK2 wildtype PMGM or CALR thrombocythaemia and various clinical stages (C1, C2, C3) with sequential stages of normocellular CALR thrombocythaemia (M) and hypercellular thrombocythaemia (MG) related to the degree of myelofibrosis.
CLM: Clinical, Laboratory, and Molecular; Hb: haemoglobin; PV: polycythaemia vera; CML: chronic myeloid leukaemia; PMGM: primary megakaryocytic granulocytic myeloproliferation; MDS: myelodysplastic syndrome; EMG: erthrocytic megakaryocytic granulocytic; RF: reticuline fibrosis; MF: myelofibrosis; LDH: lactate dehydrogenase.
Following sequencing and mutation screening in a cohort of 896 MPN patients, CALR mutations were detected in 78 of 311 (25%) ET patients, in 72 of 203 (35%) PMF patients, and in none of 382 PV patients. A total of 36 types of somatic CALR mutations (insertions and deletions) caused a frameshift reading frame with the resulting mutant CALR protein that shares a novel sequence in exon 9 with the C-terminal becoming positively charged amino acids, whereas the C-terminal of non-mutant CALR protein is negatively charged. Mutations of Type 1 (52-bp deletion) and mutations of Type 2 (5bp-insertions) accounted for 53% and 31.7% of all CALR cases. Other CALR variant mutations were observed at low frequencies or only in a single JAK2 wildtype ET or MF patient.
A large cohort of 1,235 ET and PMF patients carried the JAK2V617F, MPL515, and CALR exon 9 mutation in 63.4%, 4.4%, and 23.5% of cases, respectively, and 8.8% were triple negative for these clonal markers.22 CALR mutations mutually excluded both JAK2V617F and MPL515 mutations since all CALR mutated ET and MF patients were negative for JAK2V617F, exon 12 JAK2, and MPL mutations. The CALR mutation was detected in 195 of 289 (67%) JAK2/MPL wildtype ET, and in 105 of 120 (80%) JAK2/MPL wildtype MF. The CALR mutation was found in none of the 45 chronic myeloid leukaemia patients, 73 of myelodysplastic syndrome patients, 64 of chronic myelomonocytic leukaemia patients, and in 3 of 24 refractory anaemia with ringed sideroblasts associated with marked thrombocytosis (RARS-T) patients. The 24 RARS-T patients carried the JAK2V617F in 10 cases, MPL in 2 cases, CALR in 3 cases, and SF3B1 in 16 cases.22 The UK MPN study Group of Dr Tony Green and co-workers detected the CALR somatic mutation in 110 of 158 JAK2/MPL wildtype MPN samples (80 of 112 ET, and 18 of 32 MF samples) in none of 511 JAK2V617F or exon 12 JAK2 mutated MPNs and in 10 of 120 myelodysplastic syndrome samples: RA in 5 of 53 cases, RARS in 3 of 27 cases, RA with excess blasts in 2 of 17 cases, CMML in 1 of 33 cases, and atypical CML in 1 of 29 cases.23 The somatic CALR mutation was not found in 502 solid tumours, 1,015 cell lines, and 505 controls.23
The 52-bp deletions (CALR Type 1) eliminate almost all negatively charged amino acids, whereas the 5-bp insertions (CALR Type 2) retain aproximately half of the negatively charged amino acids.22 Such genetic differences in Type 1 and Type 2 CALR mutations predict different clinical phenotypes. CALR Type 1 deletions occur more frequently in MF than in ET.22 The USA–Italian study of Tefferi and Vanucchi24 divided 1,027 ET patients into a test (n=402) and validation cohort (n=625). Among 402 ET patients, 227 (57%), 11 (3%), and 114 (28%) harboured JAK2, MPL, and CALR mutations, respectively and 12% were triple negative.24 The 114 CALR ET patients were Type 1 in 51 (45%) and Type 2 in 44 (39%). Male sex was associated with Type 1, younger age with Type 2 variants, and platelet counts were significantly higher in Type 2 versus Type 1 CALR ET in the test and validation (n=111) cohorts of CALR ET patients.24 A large French study by Cabagnols et al.25 of 368 CALR MPN patients analysed the association of CALR Type 1 and Type 2 in ET (n=251) and MF (n=64) patients. The ratio of CALR ET to MF patients was 3.9.25 The relative frequency of CALR Type 1 versus CALR Type 2 in 251 ET patients was 51% versus 39% and in 64 MF patients it was 70% versus 13%; the median age was 61 years in Type 1 and 52.5 years in Type 2 patients; and the mean platelet count was 731 x109/L in CALR Type 1 and 870×109/L in Type 2 CALR MPN patients.25 A higher allelic burden was more frequent in CALR MF (5/35=14.3%) than in ET (6/158=3.8%). CALR MPN patients with a low allelic burden (<25%) were only observed in ET (19/158= 11.9%). CALR ET and MF patients were younger and had higher platelet counts than JAK2 ET patients in several studies.22,23-26 Leukocyte alkaline phospatase scores in the recent study of Kondo et al.27 was normal to decreased in CALR MPN. Increased LAP scores are a prominent feature of JAK2V617F, ET, PV, and masked PV.4,21,28
THROMBOSIS FREE AND OVERALL SURVIVAL IN JAK2, CALR, AND MPL MUTATED MYELOPROLIFERATIVE NEOPLASMS
In the original Austrian study by Kralovics of 186 CALR, 576 JAK2, and 35 MPL mutated ET patients, the overall survival (OS) at 10 years was 96.9% for CALR ET patients and 91.1% in JAK2V617F ET patients.29 In the Italian ET study of 89 CALR, 369 JAK2V617F, and 25 MPL515 mutated and 93 wildtype ET patients, the frequencies of microvessel symptoms were 24.7%, 27.4%, 56%, and 21.5%, respectively,30 whereas the frequencies of major thrombosis at diagnosis, in the preceding 2 years and during follow-up were 13.5%, 30.1%, 40.0%, and 16% in CALR, JAK2, MPL, and wildtype ET patients, respectively.30 The major thrombosis free survival was significantly longer in 89 CALR and 93 wildtype patients as compared with 369 JAK2 and 25 MPL ET patients. The cumulative incidence of major thrombosis at 10 years were 5.1%, 14.5%, 19.5%, and 8.1% in CALR, JAK2, MPL, and wildtype thrombocythaemia patients, respectively.30 In contrast, the Kaplan-Meier OS curves did not show significant differences between JAK2, MPL, and CALR ET patients, which indicated that the high frequency of major thrombosis patients in JAK2 and MPL ET as compared to CALR ET patients has no prognostic significance in terms of OS in ET.30
CALR MF patients had a better OS than JAK2 MF patients than in CALR MF (p=0.049) in several studies.22-26 In the Austrian study, 98 CALR MF patients had a longer OS (median: 21 years) as compared with 189 JAK2 MF (median: 11.0 years) and 18 MPL MF patients (median: 8.2 years).29 The OS curves from a large Italian PMF study)29 in 72 CALR, 396 JAK2, 25 MPL, and 53 triple negative MF patients show that CALR MF patients had a better median OS than JAK2 MF patients (hazard ratio: HR CALR/JAK2 2.3, p<0.001), MPL-MF patients (HR MPL/JAK2 2.6, p=0.009) and triple-negative MF patients (HR wildtype/JAK2 6.2, p<0.001).29 The median ages at time of diagnosis were 50, 63, 64, and 67 years for CALR, JAK2, MPL, and triple negative MF patients, respectively, indicating that the ultimate median age reached at time of death from MF will be near to 75 years and quite similar in PMF patients caused by the driver mutations CALR, JAK2, and MPL for MPN.22,29
CLINICAL, LABORATORY, MOLECULAR, AND PATHOLOGICAL FEATURES OF MYELOPROLIFERATIVE NEOPLASMS
The large cross-sectional Korean study of 407 WHO-defined MPN patients (111 PV, 179 ET, and 117 MF)31 translated the 2016 WHO classes of ET, PV, and MF patients into four distinct CLMP classes of JAK2V617F, exon 12 JAK2, MPL515, and CALR myeloneoproliferations. The three driver mutations were detected in 82.6% of 407 MPN patients and showed a distribution frequency of three distinct MPNs: JAK2 in 275 patients (67.5%), CALR in 55 patients (13.7%), and MPL in 6 patients (1.5%). The clinical phenotypes in 275 JAK2 mutated MNP were PV in 101 cases, ET in 95 cases, and MF in 79 cases. The clinical phenotypes in 56 CALR mutated MPN were PV in no cases, ET in 40 cases, and MF in 16 cases.31 The clinical phenotypes in six MPL cases were ET in three and MF in three. The seven cases of exon 12 JAK2 were diagnosed as PV in its purity and none as ET or MF.31
The mean age of CALR mutated MPN patients (57.5 years) was 8.5 years younger than in JAK2 mutated MPN patients (66 years).31 Exon 12 JAK2 mutated MPN patients presented with increased erythrocyte counts >5.8×1012/L, normal platelet counts of <350×109/L, and no anaemia consistent with the diagnosis of erythrocythemic PV (Figure 2).31 CALR mutated MPN (ET and MF) patients presented with normal to decreased values for Hb, Ht, and erythrocytes (upper limit <5.8/5.6×1012/L) (Figure 2). Erythropoiesis in bone marrow histology studies was normal or reduced in all cases of CALR and MPL mutated MPN.32
The values for Hb, Ht, and erythrocyte counts in 2016 WHO-defined JAK2V617F mutated MPN cases ranged from anaemic in MF, normal in ET, and increased in PV when the CLMP criteria are applied (Tables 1 and 2). Bone marrow lineage proliferation profile in 265 WHO-defined JAK2 mutated MPN revealed monolinear megakaryocytic proliferation in 29.1% of cases; dual proliferation of erythropoiesis and megakaryopoiesis (EM, prodromal PV) in 13.5% of cases; trilinear proliferation of erythropoiesis, megakaryopoiesis, granulopoiesis (EMG, classical PV) in 31.3% of cases; and granulopoiesis megakaryopoiesis (JAK2 MF) in 26.2% of cases when the simplified and improved EuroAsian CLMP criteria in Tables 1, 2, and 3 were applied.31 Bone marrow lineage proliferation profile in 56 CALR mutated MPN cases revealed E and EG in zero, monolinear megakaryocytic in 66%, and dual GM in JAK2/MPL wildtype, but CALR mutated MPN in 34%31 (formerly diagnosed as ML by Dameshek3 or PMGM myeloneoproliferation [MNP] by Michiels et al.4).
The JAK2 allele burden in WHO defined JAK2V617F mutated MPN (ET, PV, MF) from the Korean cross- sectional MPN study31 was widely distributed from 1.8–98.6%. The allele burden in exon 12 JAK2 mutated MPN remained <50%, which is completely in line with the heterozygosity of mutated exon 12 at the EEC level.33 JAK2V617F mutated ‘forme fruste’ PV (prodromal PV), early PV, and exon 12 PV patients presented with EM bone marrow neoproliferation without fibrosis (MF 0/1).31 The allele burden in EM and EGM of the two JAK2 mutated MPNs was significantly higher in JAK2V617F (84.9%) than in exon 12 JAK2 (44.5%) MPN. The mean values of the JAK2V617F allele burden in megakaryocyte (=ET), GM (=MF), EM (=PV), and EGM (=PV) bone marrow proliferations were 37.5%, 68.9%, 76%, and 89.2%, respectively. The JAK2V617F EM and EGM molecular pathologic (MP) groups are associated with high allele burden and increased erythrocytes (>5.8×1012/L, Figure 2), which is consistent with the diagnosis of classical PV. The normocellular megakaryocytic molecular genetic-pathologic (GP) groups in various clonal MPNs are associated with normal erythrocytes and leukocytes consistent with the diagnosis JAK2 or CALR mutated thrombocythaemia and have the lowest allele mutation burden.31 JAK2V617F EGM and GM molecular GP groups have the highest allele burden and most pronounced leukocytosis, whereas allele burden and leukocytosis are much less pronounced in the CALR GM GP group. The grade of fibrosis in the Korean study31 was divided into minimal (MF 0/1) and overt (MF 2/3), according to standardised criteria.4,29,34 The frequency of overt fibrosis in JAK2V617F and CALR-mutated and triple-negative MPN patients were 22.2%, 27.1%, and 29.3%, respectively. JAK2V617F-GM and CALR-GM bone marrow histology showed a high rate of overt fibrosis (46.0 and 42.1%), followed by JAK2V616F-M (17.5%), CALR-M (17.2%), and JAK2V617F EGM (10.4%; p<0.001). None of the JAK2-EM (‘forme fruste’ and early PV and exon 12 PV) patients presented overt fibrosis. Bone Marrow Fibrosis (BMF) Grade MF 0/1 versus Grade 2/3 appeared to be a main adverse prognostic factor when associated with JAK2V617F and triple negative MPN disease.31
MOLECULAR PATHOBIOLOGY CALR THROMBOCYTHAEMIA AND MYELOFIBROSIS
All JAK2 and MPL thrombocythaemias are driven by indirect cytokine activation (JAK2V617FSTAT5 or TPOTpoR=MPL), or direct cytokine activation (MPL515 and MPL505) cytokine receptor activation. Constantinescu, Kralovics, and Vainchenker measured STAT5 transcritional activity in CALR mutated and wildtype Ba/F3 cells along with a cytokine receptor TpoR, EpoR, and GCSFR.32 CALR mutants Type 1 and 2, but not wildtype CALR, did induce STAT5 activation via TpoR (MPL) and GCSFR, but not via EpoR, and not by CALR mutants lacking exon 9. The STAT5 activation via GSCFR was much weaker than via TpoR (MPL). CALR mutants Type 1 and 2 could not induce TpoR (MPL) activation in the absence of the JAK2 gene. In transiently HEK293 cells, CALR mutants induced dimerisation of JAK2 in the presence of TpoR (MPL), but not of EpoR. The extracelllar domain of TpoR (MPL), but not of EpoR, was indispensible for CALR mutant induced activity and the D1D2 distal part of the extracelluar TpoR domain and its associated N-glycosylation sites but not the TPO binding site in the D3D4 domain of TpoR control CALR mutant activity, which was more pronounced for CALR del52 (Type 1) than for CALR ins5 (Type 2) mutants. The Asn residues N117 and N178 present in D1D2 are key players in TpoR (MPL) activation by CALR mutants. Knocking down either MPL/TpoR or JAK2 in megakaryocytic progenitors from CALR thrombocythaemia patients inhibited cytokine-independent (spontaneous) megakaryocyte colony formation. Using a retrovirus mouse bone marrow transplant model clearly showed the induction of an MPL-mediated thrombocythaemia in CALR mutated mice.35 CALR del52 Type 1 mutation and, to a lesser extent, CALR ins5 Type 2 mutation induced thrombocythaemia due to megakaryocytic myeloneoproliferation in the early post-bone marrow transplant period. The CALR-thrombocythaemia disease was transpantable into secondary recipients. After 6 months, CALR del2 Type 1 thrombocythaemia mice, in contrast to rare in CALR ins5 transduced mice, developed a MF phenotype associated with splenomegaly and marked osteosclerosis mimicking the natural history of CALR thrombocythaemia into MF, myeloid metaplasia of the spleen, and hypocellular MF in patients with JAK2/MPL wildtype PMGM (Table 3).20,21
Araki et al.36 found that expression of CALR mutants in UT-7/TPO and YT-7/EPO cells induces TPO independent growth of UT-7/TPO cells but not of UT-7/EPO cells. C-MPL (TpoR) is required for this TPO-independent growth of UT-7/TPO cells. The CALR mutant specific carboxyterminal terminus portion (D1D2) binds to the P-domain of the CALR mutant to allow the N-domain of the mutant CALR to interact with c-MPL (TpoR), thereby explaining the gain-of function activity of CALR mutants Type 1 and 2. CALR mutants activate the JAK2 downstream pathway via binding to c=MPL (TpoR) in UT-7/TPO cells and in TOP-independent megakaryopoiesis in induced pluripotent stem cells and this induction was blocked by JAK2 inhibitors.
CONCLUSION
The cross sectional Korean MPN research study on GP characteristics31 could translate the 2016 WHO classification13 into a new set of improved EuroAsian CLMP criteria for the diagnosis and staging of MPN (Tables 1, 2, and 3).37 The 2017 CLMP criteria will pick up asymptomatic latent, masked, early stage, and symptomatic overt stages of thrombocythemias and polycythaemias 5–10 years earlier compared to the 2008—2016 WHO classifications. Prefibrotic JAK2V617F normocellular thrombocythaemia, prodromal PV, and the sequential stage of classical PV and masked advanced PV as well as prefibotic normocellular MPL thrombocythaemia and CALR thrombocythaemia ET in the complete absence of any signs of PV are poorly or not defined by the 2016 WHO classification.13,37 The EuroAsian CLMP criteria in Tables 1, 2, and 3 are based on detailed analysis and interpretation of recent advances in the molecular aetiology and pathobiology of JAK2 trilinear MPN, exon 12 PV, MPL thrombocythaemia, and CALR thrombocythaemia and MF, which have important prognostic and therapeutic implications.37 The diagnostic differentiation staging related to the natural history of prefibrotic (MF 0/1) and fibrotic (MF 2/3) JAK2, MPL, and CALR mutated MPNs should be based on bone marrow megakaryocyte morphology, bone marrow cellularity due to increased erythropoiesis and/or granulopoiesis, JAK2, MPL, and CALR mutation load, and the degree of anaemia, bone marrow fibrosis, and splenomegaly.32,37





