Interview Summary
The thalassaemias are a heterogeneous group of inherited chronic blood disorders associated with impaired haemoglobin (Hb) synthesis, resulting in ineffective erythropoiesis, haemolysis, and the development of lifelong anaemia. The pathophysiology of thalassaemia is due, in part, to increased energy demand to clear globin aggregates and reactive O2 species, and maintain overall red blood cell (RBC) health, coupled with insufficient adenosine triphosphate (ATP) production.
For this article, interviews were conducted by EMJ in December 2022 with two key opinion leaders. Kevin Kuo is a Clinician-Investigator and Staff Haematologist at the University Health Network, Toronto, Canada, and Associate Professor in the Division of Hematology, Department of Medicine, University of Toronto, Canada. Maria Cappellini is Professor of Medicine in the Department of Clinical Sciences and Community, University of Milan, Italy. Both haematologists have over 50 years of expertise and clinical experience between them in treating patients with thalassaemia. The two experts provided insights into two ongoing Phase III clinical trials for both α- and β-thalassaemia. These trials are investigating the effect of mitapivat, a first-in-class, small molecule pyruvate kinase activator, in patients across the full range of thalassaemia subtypes. The ENERGIZE trial is investigating mitapivat in patients with non–transfusion-dependent thalassaemia (NTDT), and the ENERGIZE-T trial is investigating mitapivat in patients with transfusion-dependent thalassaemia (TDT).
OVERVIEW OF THE RANGE OF THALASSAEMIA SUBTYPES
The thalassaemias are a heterogeneous group of hereditary blood disorders characterised by an absence or reduced synthesis of either α- or β-globin chains, which are the two major proteins of Hb, the O2-carrying component of RBCs.1 There are several phenotypes of thalassaemia that are typically grouped as either α- or β-thalassaemia, with α-thalassaemia characterised by reduced or absent synthesis of α-globin chains, and β-thalassaemia by reduced or absent β-globin chains.1 This causes an excess of unpaired α- and β-globin chains, producing an imbalance, and leading to excess globin aggregates that form toxic, insoluble precipitates in RBCs. This imbalance results in the impaired synthesis of Hb, leading to ineffective erythropoiesis (inability for RBCs to reach maturity), haemolysis (RBC rupturing), and inefficient RBC metabolism, including a reduced production of ATP.2 Cappellini noted that there are “a range of subtypes from homozygous to heterozygous with over 400 mutations seen in the β-globin chain alone.” The severity of thalassaemia can range from mild to severe, depending on the number and type of gene mutations.1
Cappellini explained that there are heterozygous subtypes within α- and β-thalassaemia, including α- or β-thalassaemia minor, which can result in mild microcytic anaemia that may not require clinical management.1 The more severe β-thalassaemia is homozygous β-thalassaemia major, which is caused by the complete absence of β-globin and is also known as β-zero thalassaemia, resulting in transfusion dependency.1 Another type of thalassaemia involving HbE results in HbE-β-thalassaemia, which can have a range of clinical severity.1 Patients with β-thalassaemia intermedia may present later in life and exhibit mild-to-moderate anaemia with varying transfusion needs.1 When there is a complete absence or severe deficiency of α-globin, this results in α-thalassaemia major, or Hb Bart hydrops fetalis syndrome, which is frequently fatal;1 whereas α-thalassaemia intermedia, or HbH disease, presents with haemolytic anaemia, with an Hb between 7–10 g/dL, and hepatosplenomegaly.1 Patients with α-thalassaemia intermedia may only require occasional blood transfusions, for example, during pregnancy or infection, although transfusion requirements can increase with age.1
Cappellini highlighted that ineffective erythropoiesis and the inability to maintain Hb homeostasis “places stress on the bone marrow, resulting in reduced survival and differentiation of RBC progenitors, meaning a reduction in the number of mature RBCs in circulation.” The result is the development of haemolytic anaemia, which can lead to the need for life-long blood transfusions.2,3 Kuo stated that the need for blood transfusions is determined by a low Hb level, as well as consequences related to ineffective erythropoiesis, haemolysis, and anaemia, such as failure to thrive, growth and pubertal delays, bone marrow expansion, extra-medullary haematopoiesis, and symptomatic splenomegaly.1,4
Kuo and Cappellini outlined that currently, the global standards of care for thalassaemia are inequitable, due to the variation in treatment availability, the excess burden of disease in some regions, inaccessibility of safe blood supplies, the lack of availability of affordable treatment options, and the varied quality of clinical care infrastructure.5,6
The following interview from two experts, Kuo and Cappellini, presents an overview of the Phase III ENERGIZE and ENERGIZE-T clinical trials that are investigating the effect of mitapivat, a pyruvate kinase activator, for both TDT and NTDT α- and β-thalassaemia.
Transfusion-Dependent Thalassaemia and Non-Transfusion-Dependent Thalassaemia
Chronic anaemia reflects the pathophysiology of thalassaemia and with more severe anaemia (Hb level: <7 g/dL) there is a reliance on blood transfusions, hence the categorisation of TDT compared to NTDT.7 Kuo explained that the standard of care for severe β-thalassaemia is regular transfusions, every 2–4 weeks. Transfusions are normally initiated between >4 months–4 years of age, when indicated by failure to thrive and an inability to sufficiently maintain Hb levels. Changes in transfusion requirements (i.e., progressing from NTDT to TDT), typically occur during puberty as metabolic demands increase. For α-thalassaemia NTDT, this does not follow a standardised treatment approach of regular transfusions, and in HbH disease, the transfusion requirement is more sporadic. Phenotypes such as HbH disease are associated with chronic haemolytic anaemia and clinical sequelae, where a Hb level of <10 g/dL defines patients at risk of developing morbidity and is associated with significantly shorter morbidity-free survival.6-9
Regarding thalassaemia treatment, Cappellini summarised that the “key point is to try to correct the chronic anaemia, because that will improve the outcome of general well-being, but also will impact on ineffective erythropoiesis.”
ENERGIZE AND ENERGIZE-T PHASE III CLINICAL TRIALS IN α- AND β-THALASSAEMIA
Chronic Hb levels of <10 g/dL have demonstrated an increased association with serious comorbidities.9,10 Therefore, treatment options that improve anaemia and RBC health may prevent the development of thalassaemia-related comorbidities, especially in younger patients, and circumvent the need for early intervention to prevent consequences associated with anaemia. Cappellini said that even in TDT, an increase in Hb may decrease transfusion burden, risk of iron overload, and the need for chelation therapy. Therefore, potential agents that target ineffective erythropoiesis to address the anaemia are of particular interest.11
Proposed Mechanism of Action of Mitapivat in Thalassaemia
Mitapivat is a small molecule pyruvate kinase activator (including activation of the key RBC isoenzyme, pyruvate kinase-R), which is involved in the last stage of the glycolytic pathway, activating pyruvate kinase, thus converting phosphoenolpyruvate into pyruvate to generate ATP for cellular function.12-14 As mature RBCs lack a nucleus or mitochondria, these cells are dependent on glycolysis as the essential energy pathway for generating ATP.
Kuo highlighted that studies have shown a paucity of ATP in RBCs in patients with β-thalassaemia.15 They identified that, in thalassaemia, an imbalance of α- or β-globin chains leads to the aggregation and precipitation of excess globin that results in the generation of reactive O2 species. In turn, the increased oxidative damage causes haemolysis and ineffective erythropoiesis, leading to anaemia and subsequent iron overload due to dysregulated iron metabolism.16
Mitapivat was investigated in a Phase II proof-of-concept trial conducted at study sites in Canada, the UK, and the USA in 2018–2020, the results of which were published in 2022.17 Kuo stated that the study demonstrated an acceptable safety profile, saying that “16 of 20 patients (5 out of 5 α-thalassaemia and 11 out of 15 β-thalassaemia) had a ≥1.0 g/dL increase in Hb concentration from baseline at one or more assessments between Weeks 4–12.” Most adverse events were of mild to moderate severity. The most commonly reported adverse events were initial insomnia (defined as the inability to fall asleep), dizziness, and headache.17 The results from the Phase II study supported the initiation of the ENERGIZE18 and ENERGIZE-T19 Phase III clinical trials. The safety and efficacy of mitapivat in thalassaemia is still under investigation and has not been established. There is no guarantee that mitapivat will receive health authority approvals or become commercially available in any country for thalassaemia.12-14
The Design of the ENERGIZE and ENERGIZE-T Phase III Clinical Trials
Both the ENERGIZE and ENERGIZE-T Phase III clinical trials are double-blind, randomised, placebo-controlled, multicentre studies.18,19 The studies are evaluating the efficacy and safety of mitapivat versus placebo in adult patients (18 years and above) with a diagnosis of α- or β-thalassaemia, including β-thalassaemia with or without α-globin gene mutations, HbE-β-thalassaemia, or α-thalassaemia HbH disease. Those patients with NTDT were recruited to the ENERGIZE trial and those with TDT were recruited to the ENERGIZE-T trial. The double-blind period for the ENERGIZE trial is for 24 weeks, and the ENERGIZE-T trial is for 48 weeks.18-20
Kuo, who is the global principal investigator for the ENERGIZE trial and a co-investigator for the ENERGIZE-T trial, noted that these trials were novel, incorporating both patients with α- and β-thalassaemia, and stated that there is “no specific genotypic exclusion, as long as patients have a clinical diagnosis of thalassaemia.” Cappellini emphasised the importance of the inclusion of both patients with α- and β-thalassaemia in these clinical trials, as the mechanism of action of mitapivat may prove efficacious across the full range of patients with thalassaemia. Kuo stated that “we are targeting an effect that is common to both α- and β-thalassaemia, which is the oxidative stress imposed on the RBCs,” and that mitapivat’s mode of action is “indiscriminate of the [thalassaemia] genotype.”
Both trials include an open-label extension period in which eligible patients can transition to receive mitapivat for up to an additional 5 years after the double-blind period.18-20 In both clinical trials, for those participants taking hydroxyurea, the dose must be stable for ≥16 weeks before randomisation. Key exclusion criteria include, but are not limited to: a documented history of homozygous or heterozygous sickle Hb or HbC; prior exposure to gene therapy or prior bone marrow or stem cell transplantation; or, having currently received treatment with luspatercept or hematopoietic stimulating agents (with the last dose having been administered ≥18 weeks before randomisation for the ENERGIZE trial and ≥36 weeks before randomisation for the ENERGIZE-T trial).18,19
Study Locations of the ENERGIZE and ENERGIZE-T Trials
Historically, thalassaemia was regarded as highly prevalent in malaria-endemic regions.4 However, the epidemiology of thalassaemia is changing, with patients being diagnosed globally due to population migration, including in Europe and North America.4,5,7 Currently, the β-thalassaemias are more commonly seen in Mediterranean, Middle East, and South Asian countries, as well as the Indian subcontinent, with α-thalassaemias being more prevalent in South East Asia and the Middle East (Figure 1).4,5,7
Participant recruitment for both the ENERGIZE and ENERGIZE-T trials were based on a clinical diagnosis of thalassaemia, with enrolment taking place in several regions, including those countries seeing an increasing prevalence of thalassaemia based on migration. Study sites are based in 20 countries across the globe in Brazil, Bulgaria, Canada, Denmark, France, Germany, Greece, Italy, Lebanon, Malaysia, the Netherlands, Saudi Arabia, Spain, Taiwan, Thailand, Türkiye, the United Arab Emirates, the UK, and the USA (Figure 1).18,19
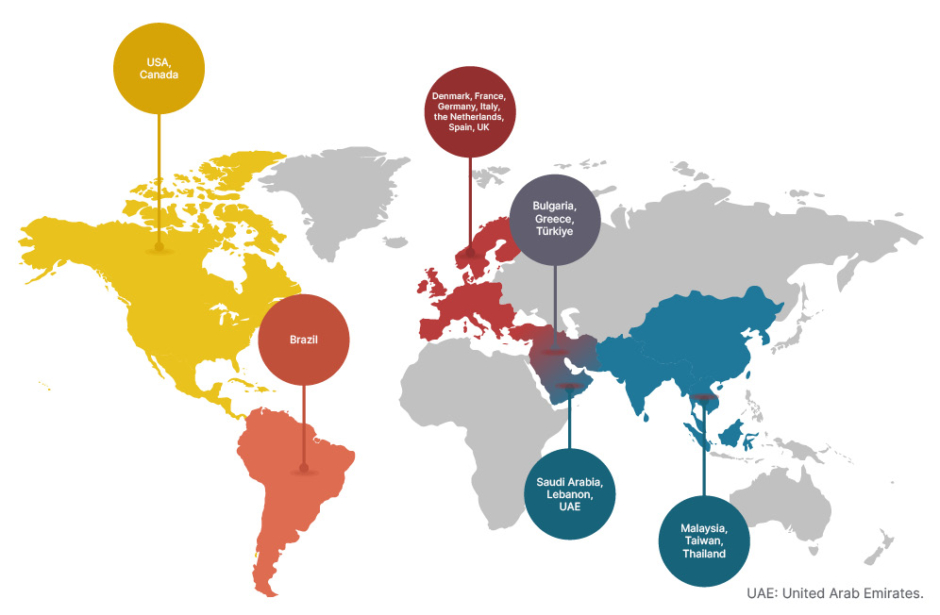
Figure 1: Geographical distribution of thalassaemia-prevalent countries including non-endemic migrant regions. Coloured pins denote the location of the countries where study sites are involved in the ENERGIZE18 and ENERGIZE-T19 Phase III clinical trials.
ENERGIZE Study in Non-Transfusion-Dependent α- and β-Thalassaemia
The ENERGIZE Phase III clinical trial enrolled patients with α- and β-NTDT, with an expected sample size of 171 participants, randomised 2:1 to mitapivat or placebo (with approximately 114 and 57 patients in the respective treatment arms). Participants will receive 100 mg of oral mitapivat (or placebo) twice daily for 24 weeks (Figure 2).18,20 Non-transfusion-dependent is defined as ≤5 RBC units during the 24-week period prior to randomisation, and no RBC transfusions 8 weeks prior to the screening period or during the screening period (up to 6 weeks; up to 14 weeks in total). For inclusion in the trial, Hb concentration must be ≤10 g/dL during the screening period, based on an average of at least two Hb concentration measurements (separated by ≥7 days).18,20
Figure 2: Study design for ENERGIZE Phase III clinical trial for adults with α- or β-non-transfusion-dependent thalassaemia.18,20
Adapted from Kuo et al.20
BID: twice daily; Hb: haemoglobin.
The primary endpoint is Hb response, which is defined as a ≥1.0 g/dL increase in average Hb concentration from Week 12 through Week 24 compared with baseline.18 Kuo stated the importance of Hb as an endpoint as “there is a cumulative increase in risk and the number of complications with progressively lower Hb.”6
A key secondary endpoint is fatigue, determined by the Functional Assessment of Chronic Illness Therapy (FACIT)-Fatigue subscale scored from Week 12 through Week 24, which reflects patient-reported outcomes.18 Kuo highlighted the importance of recognising patient-reported outcomes in the current clinical trial with the inclusion of tools such as FACIT, which reflect endpoints that are functionally relevant to patients. Additional secondary outcomes include haematological markers such as changes in Hb concentration from Week 12 through Week 24; markers of haemolysis and ineffective erythropoiesis; improvements in the Patient Global Impression of Severity (PGIS) and Patient Global Impression of Change (PGIC) ratings; distance achieved in the 6-minute walk test; and adverse events.18,20
ENERGIZE-T Study for Transfusion-Dependent α- and β-Thalassaemia
The ENERGIZE-T Phase III clinical trial enrolled patients with α- and β-TDT, where transfusion-dependent is defined as 6–20 RBC units transfused and no transfusion-free period greater than 6 weeks during the 24-week period prior
to randomisation.19,20
The trial has an expected total sample of 240 participants, with a 2:1 randomisation to mitapivat or placebo (approximately 160 and 80 patients, respectively). In the ENERGIZE-T trial, participants receive 100 mg of oral mitapivat (or placebo) twice daily for 48 weeks (Figure 3).19,20
Figure 3: Study design for ENERGIZE-T Phase III clinical trial for adults with α- or β-transfusion-dependent thalassaemia.19,20
Adapted from Kuo et al.20
BID: twice daily; RBC: red blood cell.
The primary endpoint is the proportion of patients with a transfusion reduction response, defined as ≥50% reduction in transfused RBC units, with a reduction of ≥2 units of transfused RBCs in any consecutive 12-week period through Week 48 compared with baseline. Secondary outcomes include additional assessments of transfusion burden and markers of iron metabolism.19,20
Cappellini indicated that, from their clinical experience, “patients only identify the impact of anaemia on their wellbeing and capabilities when the anaemia is improved with treatment, when they realise that they can do more and be more active than they are used to.” Kuo stated that their goal was “to examine the long-term effects of mitapivat in this population.”
FUTURE PROSPECTS
The current Phase III ENERGIZE and ENERGIZE-T trials are ongoing. Kuo indicated that the results of the trials are likely to become available in the next 2 years. Pending the results from ENERGIZE and ENERGIZE-T, paediatric trials in thalassaemia are also planned. Both the ENERGIZE and ENERGIZE-T studies are pivotal in assessing mitapivat as a potential treatment for haemolytic anaemia across a broad range of patients with α- and β-thalassaemia subtypes, with TDT and NTDT.





