
Abstract
There are ˜3,000 children, as well an additional ˜7,000 adults, diagnosed with acute lymphoblastic leukaemia (ALL) each year in the USA. This makes ALL the most common cancer diagnosed in children. It represents ˜25% of paediatric cancer diagnoses. With current therapy, most patients achieve a complete remission and many are cured. However, the prognosis remains quite poor for the ˜15–20% of children who suffer a relapse of their ALL. Improved outcomes for these relapsed patients will require either more efficacious salvage therapies or improved initial therapy that prevents ALL relapse. Thus, understanding the mechanisms by which a small population of leukaemia cells can escape therapy and contribute to relapse often months or years later is critical for improving ALL outcomes. Herein, we will review emerging clinical and laboratory research that suggest quiescence, or dormancy, is an important cellular mechanism that enhances ALL chemo-resistance and persistence, and ultimately contributes to disease relapse. Furthermore, the mechanisms that regulate this balance between leukaemia quiescence and proliferation are beginning to be elucidated and will provide new knowledge about leukaemia biology. Finally, these observations support the need for and feasibility of therapeutically targeting these quiescent, chemo-resistant ALL cells by either exploiting metabolic or signalling pathway vulnerabilities unique to quiescent cells, or by causing the release of ALL cells from the protective niche(s) that triggers and maintains ALL quiescence.
INTRODUCTION
Acute lymphoblastic leukaemia (ALL) is the most common childhood malignancy, accounting for ˜25% of all paediatric cancer diagnoses.1 Despite significant advances in leukaemia therapy, ˜15–20% of paediatric patients experience disease relapse months or years after achieving a complete remission.2 Relapsed leukaemia is typically refractory to conventional therapy and portends a poor prognosis.3,4 The mechanisms by which a small subset of leukaemia cells escape therapy and persist at undetectable levels for extended periods of time before resuming proliferation are not completely understood. Identifying these mechanisms of leukaemia relapse is critical for developing more efficacious therapies that fully eradicate the disease.
Leukaemia cell quiescence, or dormancy, is one mechanism that likely contributes to leukaemia chemo-resistance, persistence, and relapse.5 Quiescent cells are in the G0 phase of the cell cycle, such that they are neither dividing nor are they preparing to divide. Accordingly, quiescent cells differ from proliferating or ‘cycling’ cells by low RNA content and DNA replication machinery, lack of proliferation markers, and diminished metabolism. Importantly, quiescence is a reversible state from which cells may re-enter the cell cycle and proliferate in response to stimuli.6
This ability to remain quiescent for extended periods of time, while retaining the ability to self-renew and differentiate, is critical for adult stem cell biology, tissue homeostasis, and is a feature of ‘stemness’ that defines these cells.7 For example, a small fraction of quiescent haematopoietic stem cells (HSC) ensure a lifelong supply of mature blood cells as well as maintain haematopoiesis in the face of catastrophic haematopoietic cell loss such as haemorrhage or exposure to cytotoxic chemotherapy.8,9 Moreover, defects in the regulation of quiescence in HSC lead to premature exhaustion of the HSC pool and haematological failure.10-13 Extensive research has shown that HSC quiescence is maintained and regulated by a complex network of HSC-intrinsic mechanisms and extrinsic signals.9,14 Quiescent HSC localise to specialised bone marrow niches, or microenvironments, largely separate from active, cycling HSC and committed, maturing progeny. It is the complex interactions between these cells and their niche that drive the balance between quiescence and proliferation.15,16 Quiescence is also a necessity among more differentiated haemapoietic cell lineages. For example, naïve T and B lymphocytes utilise quiescence to minimise the cellular energy expenditures needed to maintain an organism’s vast repertoire of lymphocytes, given that the majority of these cells will go unused. Diminished metabolism and lack of replication cycles also reduces potential metabolic damage or genetic mutations that could lead to malignant transformation.17,18
THE ROLE OF QUIESCENCE IN CANCER BIOLOGY
It is widely recognised that both solid and haematologic malignancies are not composed of a homogeneous cancer cell composition, but rather a heterogeneous mixture of cells in various stages of differentiation.19-21 Some cancers have identifiable tumour-initiating cells with stem cell-like properties.22,23 These aptly named cancer stem cells (CSC) share characteristics with normal stem cells, including the capacity to become quiescent.24,25 CSC in solid tumours, such as carcinomas, exploit quiescence to i) evade chemotherapy agents that target rapidly proliferating cells, ii) evade immune recognition, and iii) elicit neo-angiogenesis. Furthermore, CSC quiescence may also explain the delay in growth of tumour cells deposited at metastatic sites.26,27
Similar to many solid tumours, acute myeloid leukaemia (AML) is organised as a heterogeneous hierarchy that originates with a rare, immunophenotypically distinct population of leukaemia stem cells (LSC).23,28-30 While quiescence is not a universal characteristic of all AML LSC, it is one property that allows some LSC to evade the effects of anti-proliferative chemotherapy agents.31 Quiescence may also render AML LSC insensitive to drugs targeting metabolism or signalling pathway inhibitors given their limited metabolic activity and restricted intracellular signalling.5 Subsequent cell cycle re-entry and proliferation of these quiescent leukaemia cells that escape the effects of therapy can lead to cancer recrudescence months to years after initial presentation.
THE ROLE OF QUIESCENCE IN ACUTE LYMPHOBLASTIC LEUKAEMIA
In contrast to AML, studies in ALL have not conclusively identified an immunophenotypicallydefined LSC population.20,32 For example, le Viseur et al.33 showed that multiple pre-B ALL blast populations, isolated by flow cytometry and expressing a range of differentiation markers, could engraft immunocompromised mice, produce a heterogeneous leukaemia population with a range of maturation phenotypes, and give rise to the same leukaemia population diversity in subsequent transplantations. These and other results suggest a variety of different ALL subpopulations display stemness, such that they can repopulate leukaemia when transplanted into immunocompromised mice.34,35 This lack of an immunophenotypicallydefined LSC population in ALL makes it more challenging to study the contributions of quiescence to ALL biology, as there is no distinct ALL subpopulation upon which to focus investigations. Despite this difference with AML and associated challenges, clinical observations and emerging experimental data support that quiescence plays a critical role in ALL biology and clinical behaviour (Figure 1A).36-42
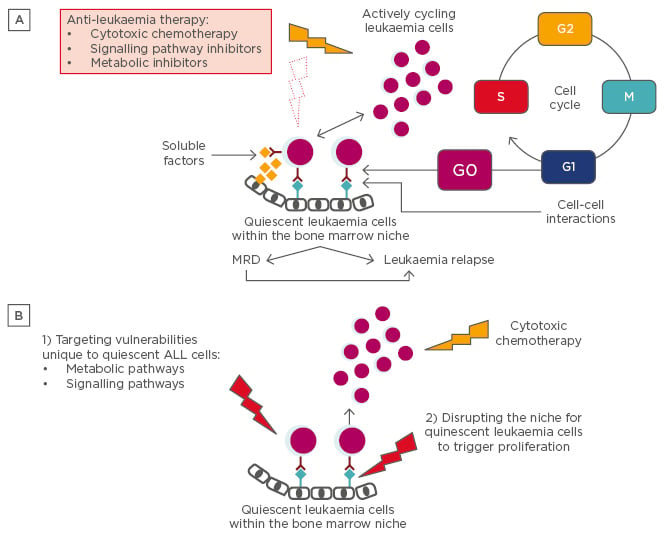
Figure 1A: Consequences of ALL quiescence. Clinical observation and leukaemia xenotransplantation experiments that allow tracking of quiescent ALL cells suggest that a small fraction of leukaemia cells reside in a protective bone marrow niche, where soluble factors and cell-cell interactions induce and maintain a quiescent state. This phenotype enables the leukaemia cells to resist conventional cytotoxic chemotherapy agents, as well as metabolic and signalling pathway inhibitors specific to more rapidly cycling cells. These cells can persist as MRD and go undetected by conventional means of staging until some future time when they re-enter the cell cycle to proliferate and cause disease relapse.
Figure 1B: Approaches for targeting quiescent ALL cells. New therapeutic targets will be identified as studies i) define how the quiescent phenotype diverges from the highly proliferative phenotype in ALL, and ii) identify factors that protect and maintain quiescent ALL cells in the bone marrow niche. For example, directly targeting vulnerabilities unique to quiescent leukaemia cells (1) or disrupting the niche for quiescent leukaemia cells to induce proliferation followed by cytotoxic agents that preferentially target proliferating cells (2) represent novel approaches to leukaemia therapy. Moreover, xenotransplantation experiments with dye-labelled leukaemia cells can be used for preclinical testing of these novel therapies.
ALL: acute lymphoblastic leukaemia; MRD: minimal residual disease.
Leukaemia quiescence is one factor that may contribute to the clinical observation that some pre-B ALL relapses can occur 5–10 years after initial presentation and presumed cure. As an extreme example of leukaemia quiescence, a patient has been described whose BCR-ABL1 pre-B ALL relapsed after 22 years of remission, subsequently giving rise to an AML immunophenotype.36 Intriguingly, the primary and relapsed leukaemia samples shared an identical BCR-ABL1 fusion sequence as well as identical immunoglobulin gene rearrangements, indicating that the relapse was a derivative of the founding clone. Further supporting a potential role for quiescence in ALL biology, the TEL-AML1 gene rearrangement, a hallmark of childhood ALL, has been identified in neonatal blood spots of leukaemia patients. This suggests that pre-leukaemic genetic changes can occur in utero and precede leukaemia by years.43 Transgenic mice expressing the TEL-AML1 fusion protein in HSC not only exhibit an increased number of HSC but also a higher percentage of HSC with a quiescent phenotype that are more prone to transformation to leukaemia.44 Additional gain and loss of function experiments with TEL-AML1 also support a role for TEL-AML1 in maintaining cellular quiescence.45,46 These, and other examples suggest that pre-leukaemic cells can persist and remain quiescent for extended periods of time.
Additional clinical evidence also suggests that quiescent ALL cells may contribute to disease relapse. Lutz et al.38 characterised the immunophenotype and cell cycle properties of the leukaemia cells responsible for minimal residual disease (MRD) present at the end of induction chemotherapy in a small cohort of paediatric pre-B cell ALL patients. At diagnosis, they identified an immunophenotypically-defined population of leukaemia cells that were more quiescent and less actively cycling than other leukaemia subpopulations. By analysing MRD leukaemia samples in the same patients, they found that chemotherapy further selected for this quiescent leukaemia population. These results support that quiescence may contribute to enhanced leukaemia chemo-resistance and MRD in ALL.
ALL xenotransplantation experiments have provided additional support for the importance of quiescence in ALL biology. Labelling of leukaemia cells with fluorescent membrane dyes prior to transplantation into mice facilitates the in vivo tracking and imaging of the leukaemia cells. An additional benefit of this approach is that the membrane dyes are retained in quiescent, non-cycling cells (dye-retaining) but diluted to undetectable levels within several cell divisions in proliferating leukaemia cells (dye-negative). Sipkins et al.39 transplanted fluorescently-labelled, human ALL cells into immunocompromised mice and then used in vivo confocal microscopy to image the leukaemia cells within the calvarium bones up to 2 weeks after transplantation. ALL cells homed to unique, spatially-restricted vascular regions within the bone marrow that expressed the chemokine SDF-1. Furthermore, these were the same microenvironments to which HSC and mature lymphocytes homed, suggesting that both benign haematopoietic cells and leukaemia cells localise to, and perhaps compete for, the same microenvironments within the bone marrow. A subsequent study by Colmone et al.40 elaborated on these findings by demonstrating distinct competition between ALL cells and HSC for the same bone marrow niche and more specifically the dysregulation of normal haematopoiesis by the establishment of the leukaemia niche.
The persistence of dye-retaining, quiescent leukaemia cells at later time points was indicative of slow cycling and potentially suggestive of quiescence in ALL cells once they entered this protective bone marrow niche. Supporting this observation, quiescent, dye-retaining leukaemia cells were identified at even later time points, including 42 days after transplantation.41 These quiescent leukaemia cells selectively localised to regions of the bone marrow expressing high levels of the glycoprotein osteopontin (OPN). OPN is secreted by endosteal osteoblasts and functions as both a soluble cytokine and an adhesive molecule within the extra-cellular matrix. Moreover, neutralisation of OPN in vivo induced leukaemia cell cycle re-entry and proliferation. Interestingly, OPN had no direct effect on leukaemia proliferation when tested ex vivo. This suggests that the in vivo effects of OPN on leukaemia quiescence may be indirect and potentially secondary to localising leukaemia cells within specific niches in the bone marrow, where additional factors trigger and maintain leukaemia quiescence. Finally, OPN neutralisation in xenotransplanted mice prior to cytarabine treatment significantly reduced the leukaemia burden relative to cytarabine treatment alone.41
Ebinger et al.42 used similar methodology with multiple pre-B and T-ALL patient-derived xenografts to further characterise ALL quiescence and therapy resistance. In agreement with the prior studies, they identified a rare population of dye-retaining, quiescent leukaemia cells 21 days after transplantation into immunocompromised mice. These quiescent leukaemia cells localised to the bone marrow endosteum, exhibited leukaemia-initiating potential upon re-transplantation, and were significantly more chemo-resistant than the proliferating, dye-negative leukaemia cells in vivo. Single cell and bulk RNA-sequencing experiments further demonstrated that these quiescent leukaemia cells exhibited a gene expression profile distinct from the dye-negative, proliferating leukaemia cells and showed high similarities to MRD cells derived from a mouse model of leukaemia, as well as MRD cells obtained from ALL patients. In addition to novel biological insights about leukaemia, these results also support the use of this experimental system as a preclinical model for testing therapeutic approaches to target MRD cells.
Finally, experiments have addressed whether quiescence represented a permanent characteristic of a subpopulation of ALL cells or rather a mutatable feature of a potentially larger number of leukaemia cells. Further supporting a reversible phenotype, when dye-negative leukaemia cells were isolated from the bone marrow of xenotransplanted mice, re-stained with a proliferation dye, and re-transplanted, they resulted in an identical dye-retaining leukaemia population as re-transplanted bulk cells. Similarly, in contrast to the in vivo results described above, quiescent, dye-retaining leukaemia cells did not exhibit increased chemo-resistance ex vivo. This draws parallels with physiological HSC quiescence and reflects a relationship between quiescence and protection within the bone marrow microenvironment.8,9,15 As described in more detail below, these results also support the feasibility of targeting these quiescent, chemo-resistant leukaemia cells by identifying approaches for removing them from their protective niche.
TARGETING QUIESCENCE AS A THERAPEUTIC APPROACH FOR ACUTE LYMPHOBLASTIC LEUKAEMIA
These studies suggest that quiescence significantly contributes to ALL chemo-resistance and disease relapse. Accordingly, directly targeting vulnerabilities unique to quiescent leukaemia cells or combining agents that disrupt leukaemia quiescence with cytotoxic agents that preferentially target proliferating cells represent novel approaches to leukaemia therapy (Figure 1B). These approaches have been more extensively investigated in the context of both HSC and AML-LSC quiescence than ALL. For example, treatment of the mice xenotransplanted with human AML cells with granulocyte-colony stimulating factor (G-CSF) triggered quiescent cells to proliferate, mobilise out of the bone marrow niche, and re-acquire sensitivity to cytarabine.47 This effect may occur, in part, through proteolytic enzyme cleavage and degradation of stem cell anchors which release the AML LSC from their protective bone marrow microenvironment.5 This strategy of combining G-CSF with cytotoxic chemotherapy in the treatment of AML has been tested in the clinic with mixed results.48-51
Several murine xenotransplantation studies have similarly demonstrated that targeting AML and CML adhesion via the CXCL12–CXCR4 axis dislodges leukaemia cells from the quiescent bone marrow niche and enhances leukaemia chemo-sensitivity.52,53 Several early-phase clinical trials have combined a CXCR4 inhibitor with cytotoxic chemotherapy in patients with AML.54-58 A theoretical concern with this approach is that HSC mobilised out of the bone marrow by CXCR4 inhibition or G-CSF stimulation may become more susceptible to chemotherapy and lead to delayed blood count recovery. Importantly, none of these trials reported prolonged blood count recovery but any added clinical benefit of this approach to standard cytotoxic AML chemotherapy still remains to be determined. However, these agents may have other effects on haematopoiesis. For example, murine experiments have shown that G-CSF treatment and CXCR4 inhibition (AMD3100) inhibit medullar B-lymphopoiesis and mobilise B-cells without affecting B-lymphopoiesis, respectively.59
The CXCL12–CXCR4 axis also plays an important role in ALL biology as murine xenotransplantation studies demonstrated that CXCR4 inhibitors, such as AMD3100 and POL5551, could reverse bone marrow mediated leukaemia chemo-resistance.60,61 Similar to the CXCL12-CXCR4 axis, the growth arrest specific-6 (GAS6)/Mer kinase axis maintains quiescence and chemo-resistance in leukaemia cells harbouring the E2A-PBX1 translocation.65 Mer kinase upregulation, driven by the E2A-PBX1 translocation, significantly stimulates secretion of its ligand, GAS6, by osteoblasts within the bone marrow niche. GAS6 subsequently promotes survival of leukaemia cells by inducing quiescence and chemo-resistance. Mer kinase inhibitors have been developed and could potentially augment the efficacy of cytotoxic chemotherapy in leukaemia patients with the E2A-PBX1 translocation and high Mer expression.66 As described above, quiescent ALL cells are localised to areas of high OPN expression in the bone marrow microenvironment. Moreover, neutralisation of OPN induced leukaemia cell cycle re-entry and proliferation as well as enhanced sensitivity to cytarabine.41 It is important to note, however, that the bone marrow niche can enhance leukaemia chemo-resistance by mechanisms independent of leukaemia quiescence.62-64 Thus, the efficacy of leukaemia-niche disrupting agents, such as CXCR4 inhibitors or other cell adhesion inhibitors, are likely multi-factorial and not solely due to effects on quiescence.
In contrast to therapeutic approaches that restore leukaemia chemo-sensitivity by disrupting the environment that promotes leukaemia quiescence, it may also be possible to identify and target vulnerabilities unique to quiescent leukaemia cells. It has been shown, for example, that quiescent AML cells are metabolically dormant and dependent on oxidative respiration rather than glycolysis for energy generation.67 Inhibition of the anti-apoptotic protein Bcl-2, which is aberrantly expressed in this leukaemia population, further impaired oxidative phosphorylation and selectively eradicated quiescent AML cells. These findings suggest it may be feasible to eradicate chemo-resistant, quiescent leukaemia populations by targeting their unique dependencies. Further supporting this possibility, several high-throughput drug screens have identified drugs that selectively target CSC, albeit not necessarily through an effect on quiescence.68-70 Finally, recent advances in immunotherapy are expanding our cancer and leukaemia therapy armamentarium.71 Unlike cytotoxic chemotherapeutics that target actively cycling cells, it is not as evident a priori that quiescent cancer or leukaemia cells will be able to escape the effects of immunotherapy. However, several studies suggest that quiescent cancer cells may evade immune therapy and surveillance by a number of different mechanisms.72-76 Thus, it is possible that approaches that disrupt leukaemia quiescence may also enhance the efficacy of immunotherapy.
CONCLUSIONS
In order to improve ALL outcomes, we must develop a better understanding of how a small population of ALL cells escape treatment and lead to relapse. Recent work has shown that ALL cells can exploit quiescence, or dormancy, to evade treatment while retaining the capacity to rapidly proliferate and differentiate. Until very recently, in vivo modelling of ALL quiescence was technically difficult due to difficulties in identifying and tracking these rare cells, but novel leukaemia xenotransplantation approaches that facilitate the tracking of quiescent ALL cells is allowing for rapid advances in the field and a more complete characterisation of the quiescent ALL phenotype. Future work is needed to define the critical cellular and molecular components of the bone marrow niche that harbour quiescent leukaemia cells as well as the leukaemia genes and pathways that induce and maintain quiescence. Isolated extra-medullary ALL relapses, such as those occurring within the central nervous system, are not uncommon and raise the possibility that other niches may also influence leukaemia quiescence and chemo-resistance.77 Similarities and differences between the leukaemia niche in the bone marrow and central nervous system are yet to be fully determined but are likely to be important for both understanding leukaemia biology and developing novel leukaemia therapies.78-81 The intrinsic and extrinsic signals that trigger quiescent leukaemia cells to re-enter the cell cycle, proliferate, and lead to disease relapse are unknown but critical for understanding the pathophysiology of relapse. It will also be critical to extend our knowledge gained from xenotransplantation models of the quiescent ALL niche to the human leukaemia niche. It is anticipated that this area of research will lead to novel ALL therapies that include targeting the metabolic and signalling pathways preferentially utilised by quiescent ALL cells as well as methods for disrupting the leukaemia bone marrow niche.





