
Abstract
Acute myeloid leukaemia (AML) is a disease with a very poor outcome and remains an area of significant unmet need, necessitating novel therapeutic strategies. The progress made in the field of immunotherapy, in particular chimeric antigen receptor (CAR)-engineered T cells, has given rise to many hopes for pathologies such as B cell acute lymphoblastic leukaemia and B cell lymphoma, and many studies have attempted to translate these successes to AML. This review summarises the recent advances in, and defines an ideal target for, CAR T cell therapy in AML.
INTRODUCTION
Acute myeloid leukaemia (AML) is an aggressive malignant disease affecting 2.5–3.5 per 100,000 adults each year in Western countries.1,2 Over the last decade, considerable advances have been made in the identification of molecular markers, leading to the improvement of risk stratification, especially in AML patients with a normal karyotype.3 However, in the past 5 years, the cure rate has been 35–40% for AML patients <60 years old and 5–15% for patients >60 years old.4 The elderly, who are unfit to have intensive chemotherapy, have a median overall survival of 4–10 months.2 Despite improved understanding of leukaemogenesis, AML still has poor outcomes due to high disease and treatment-related mortality. To date, allogeneic haematopoietic stem cell transplantation (alloHSCT) remains the most effective treatment for relapse prevention in non-favourable-risk patients with AML5,6 and has remained the most successful immunotherapeutic treatment for AML patients, especially with the advances made in using alternative donors;7 however, this therapeutic strategy has a high toxicity without guaranteeing the absence of relapse.8 Thus, it is a public health issue that new, more effective, and less toxic treatments should emerge for the treatment of AML. In this regard, chimeric antigen receptor (CAR)-engineered T cells have been developed and used in paediatric acute lymphoblastic leukaemia (ALL), resulting in complete remission without graft versus host disease (GvHD).9
CAR T cells are artificial molecules comprising an extracellular antigen-binding domain, commonly derived from a monoclonal antibody in the form of a single chain fragment variable (scFv), and an intracellular signalling domain associated with ≥1 costimulatory signalling modules.10,11 The first generation of CAR T cells were developed in ovarian metastatic carcinomas12 and built on the model scFv-spacer-CD3ζ. Unlike the T cell receptors, they recognise the antigen in the absence of major histocompatibility complex proteins; therefore, CAR T cells can specifically target various surface antigens on malignant cells.13
Nonetheless, first-generation CAR T cells exhibit a lack of cytokine secretion, insufficient proliferation, and low persistence in vivo. To improve these shortcomings, the next generation of CAR T cells involved the addition of costimulatory signalling domains, such as CD28 and 41BB or CD3ζ,14 to the cytoplasmic tail of the CAR to provide additional signals to T cells. The addition of these costimulatory proteins limited apoptosis of CAR T cells because of the synthesis of IL-2.15 Preclinical studies have indicated that this second generation improves the antitumour activity of T cells.15 Recently, third-generation CAR T cells combine multiple signalling domains, such as CD3ζ–CD28–41BB or CD3ζ–CD28–OX40, to improve activity;16 however, this improvement has not demonstrated a significant clinical impact when compared with the prior generation of CAR T cells. For example, the CAR T cell strategy is highly specific in redirecting T cells towards predefined target cells but reaches its limits when targeting solid tumours with phenotypic heterogeneity.17 After initial tumour reduction by CAR T cells, antigen-negative cancer cells not recognised by CAR may give rise to tumour relapse. This situation might be overcome by CAR-mediated activation of T cells in the tumour, releasing inducible IL-12 that increases tumour-infiltrating T cell activation and attracts and activates innate immune cells to eliminate the antigen-negative cancer cells in the targeted lesion; these fourth-generation CAR T cells are called TRUCK T cells.18 The evolution of CAR T cells is shown in Figure 1.
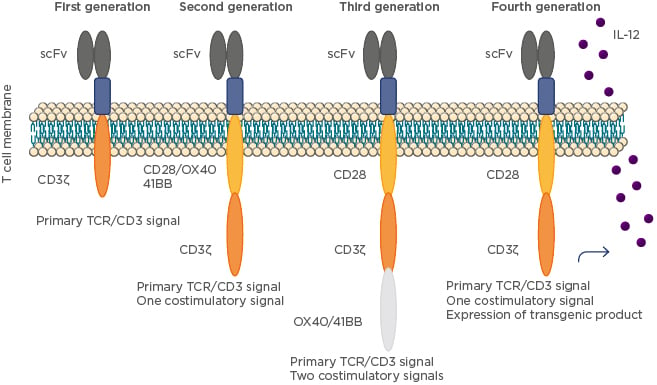
Figure 1: The evolution of chimeric antigen receptor T cells.
First-generation CAR T cells consist of a single-chain fragment of variable region antibody for target binding linked by a spacer domain to the transmembrane and intracellular signalling domain of CD3ζ derived from the T cell receptor. Addition of a costimulatory domain, mostly of the CD28 family, makes up second-generation CAR T cells. Third-generation CAR T cells contain two costimulatory domains. Fourth-generation CAR T cells (also known as TRUCK T cells) are additionally modified with a constitutive or inducible expression cassette for a transgenic protein, for instance a cytokine, which is released by the CAR T cell to modulate the tumour T cell response.
CAR: chimeric antigen receptor; scFv: single-chain fragment variable region; TCR: T cell receptor.
Recently, the U.S. Food and Drug Administration (FDA) gave approval for two third-generation CAR T cells targeting CD19;19 the first was tisagenlecleucel in B cell-lineage ALL, followed by axicabtagene ciloleucel in diffuse large B cell lymphoma and B cell-lineage ALL. This therapeutic strategy may be applicable to a large number of tumoural pathologies, including AML, for which there is a need for treatment in view of the poor results observed in adults and children. This review therefore focusses on CAR T cells for the treatment of AML.
TARGETING OF CHIMERIC ANTIGEN RECEPTOR T CELLS IN ACUTE MYELOID LEUKAEMIA
CAR T cell therapy targeting CD19 has yielded remarkable outcomes in paediatric and young adult patients with relapsed or refractory diffuse large B cell lymphoma and ALL.20-27 Many groups have attempted to reproduce these encouraging results in other pathologies, such as AML; however, because of the lack of expression of CD19 on the surface of AML cells, CAR T cells directed against CD19 have no efficacy in this pathology.28 An ideal target for CAR T cells in AML should be highly expressed in most of the malignant cells, including leukaemic stem cells (LSC), and should be shared by most AML patients. Unlike naïve T lymphocytes, which can signal through the T cell receptor in response to very low antigen density, CAR T cells need a higher density of antigen to be effective.29-32 Expression of the target by normal cells may or may not be associated with high toxicity but, to prevent toxicity, the target should not be shared by vital tissues such as the heart, liver, and normal haematopoietic stem cells (HSC). An approach to remedy this pitfall is the use of an algorithm integrating a large dataset of transcriptomics and proteomics from malignant and normal tissues to identify potential targets expressed in LSC but not in normal CD34+CD38- haematopoietic cells, T cells, or vital tissues.33 However, using CAR T cells in AML remains difficult due to the non-restricted expression of AML-associated antigens. Moreover, CAR T cells may persist in the organism for several years,34 thus an unwise target choice may lead to prolonged aplasia. Table 1 summarises the different clinical trials currently underway employing CAR T cells in AML.35-42
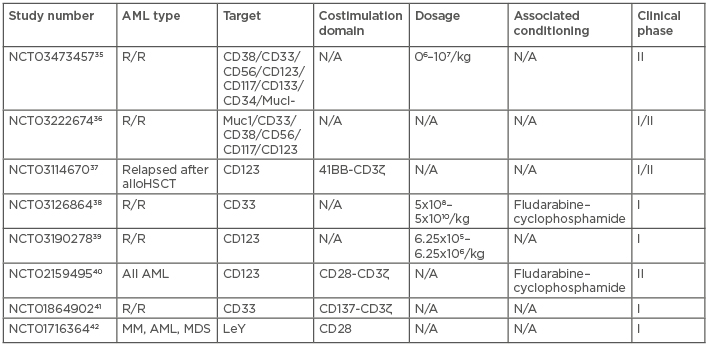
Table 1: Current clinical trials using chimeric antigen receptor T cells for acute myeloid leukaemia.
ALL: acute lymphoblastic leukaemia; alloHSCT: allogeneic haematopoietic stem cell transplantation; AML: acute myeloid leukaemia; LeY: Lewis antigen Y; MDS: myelodysplastic syndrome; MM: multiple myeloma; N/A: not applicable; R/R: relapsed/refractory.
CD33
CD33 (also known as Siglec-3) is a transmembrane receptor expressed on cells of myeloid lineage and on approximately 90% of AML cells.43-45 It is usually considered myeloid-specific but can also be found on lymphoid cells and non-haematological tissues.45 Gemtuzumab ozogamicin (Mylotarg®, Pfizer, New York City, New York, USA) is an antibody-targeted chemotherapy agent consisting of a humanised murine anti-CD33 monoclonal antibody (clone P67.6) linked to a calicheamicin Υ1 derivative via a hydrolysable bifunctional linker.46 Gemtuzumab ozogamicin induces cell death in CD33-positive cells by internalisation of the calicheamicin drug and then cleavage of double-stranded DNA,47 and has shown promising results in AML.48,49 Using an antiCD33 scFv, CAR T cells targeting CD33 have been developed for the treatment of AML, and promising results of murine AML models50,51 and humans52 have been reported. Several clinical trials are currently ongoing in AML with CAR T cells directed against CD33.
CD123
CD123 is the alpha chain of the IL-3 receptor and is a cluster-like protein of differentiation encoded by IL3RA on the human X chromosome. It is expressed on the surface of several leukaemic cells, including cells of AML, plasmacytoid,53 hairy cell leukaemia,54 and Hodgkin’s lymphoma,55 thus constituting a potential target for the treatment of these diseases. Although CD123 presents low expression levels on normal haematopoietic cells,56 it may be a possible target for AML.57 CAR T cells directed against CD123 in AML mouse models resulted in a reduction of leukaemic burden but showed contrasting results on normal haematopoiesis.58-62 Modifying the scFv directed against CD123 may be able to reduce myelotoxicity in an AML murine model,62 and clinical studies are underway in AML patients with CAR T cells targeting CD123.
Lewis Y Antigen
The Lewis antigen system is a human blood group system based on genes of chromosome 19. The Lewis antigen Y (LeY) is an oligosaccharide overexpressed on most epithelial cancer cells and haematological malignant cells, including AML cells,63,64 but is weakly expressed by healthy tissues.65,66 A Phase I study with second-generation CAR T cells directed against LeY in four AML patients reported two cytogenetic remissions without Grade 3 or 4 side effects and with long-term persistence of the CAR T cells.67 Moreover, in vivo models have shown promising results of CAR T cells targeting LeY in epithelial cancers without significant toxicity.68
FMS-Like Tyrosine Kinase 3
FMS-like tyrosine kinase 3 (FLT-3/CD135) is a receptor-type tyrosine-protein kinase, or fetal liver kinase-2, that belongs to the receptor tyrosine kinase Class III family of cytokine receptors and binds the FLT-3 ligand. It is expressed on the surface of many haematopoietic progenitor cells and its signalling is important for the normal development of HSC and progenitor cells. The FLT3 gene is one of the most frequently mutated genes in AML;69 high levels of wild-type FLT-3 have been reported for blast cells of some AML patients without FLT3 mutations. These high levels of wild-type FLT-3 may be associated with worse prognosis.70 It was recently reported that CAR T cells directed against FLT-3 in in vivo and in vitro models had a potent reactivity against AML cell lines and primary AML blasts expressing either wild-type FLT-3 or FLT-3 with internal tandem duplication. However, as anticipated, FLT-3-CAR T cells recognised normal HSC in vitro and in vivo and disrupted normal haematopoiesis in colony-formation assays, suggesting that adoptive therapy with FLT-3-CAR T cells will require subsequent CAR T cell depletion and alloHSCT to reconstitute the haematopoietic system.71
Folate Receptor β
Folate receptor β (FOLR2) is a protein encoded by the FOLR2 gene,72 and is expressed on myeloid lineage cells and frequently upregulated in AML cells.73 Preclinical models using CAR T cells directed against FOLR2 have shown promising results on AML cells while preserving normal HSC; the concomitant use of all-trans retinoic acid, which allows an upregulation of FOLR2, improved efficiency against AML.74
Hyaluronate Receptor
The hyaluronate receptor (CD44) is a cell-surface glycoprotein involved in cell–cell interactions, cell adhesion, and migration, and is expressed on many mammalian cell types. CD44 variant domain 6 (CD44v6) is a variant isoform expressed in multiple myeloma75 and AML.76 CAR T cells directed against CD44v6 have shown antileukaemic effects in mouse models, although at the cost of leukopenia.77 Further steps may involve adding a suicide gene to this construct.78
CD38
CD38, also known as cyclic ADP ribose hydrolase, is a glycoprotein79 found on the surface of many immune cells (specifically white blood cells), including T, B, and natural killer lymphocytes. CD38 also functions in cell adhesion, signal transduction, and calcium signalling.80 CD38 is expressed on AML cells but not on normal HSC.81,82 As for FOLR2, all-trans retinoic acid enhances CD38 expression and its action is synergistic with CAR T cells targeting CD38.83
Natural Killer Group 2D
Natural killer group 2D (NKG2D) is a transmembrane protein belonging to the CD94/NKG2 family of C-type lectin-like receptors.84 NKG2D is highly expressed in AML tissue but weakly on healthy tissue.85 Several CAR T cells directed against NKG2D have been clinically developed, leading to complete remissions without neurotoxicity, cytokine release syndrome (CRS), or treatment-related mortality.86
CD7
CD7 is a transmembrane protein and a member of the immunoglobulin superfamily. This protein is found on thymocytes and mature T cells and plays an essential role in T cell interactions and in T cell–B cell interaction during early lymphoid development.87 In AML, its expression correlates with chemoresistance.88 CAR T cells directed against CD7 have shown potent efficiency in vitro and in murine AML models with low toxicity.89
C-type Lectin Domain Family 12 Member A
C-type lectin domain family 12 member A (CLEC12A) is a member of the C-type lectin/ C-type lectin-like domain superfamily. Members of this family share a common protein fold and have diverse functions, such as cell adhesion, cell–cell signalling, glycoprotein turnover, and roles in inflammation and immune response. The protein encoded by this gene is a negative regulator of granulocyte and monocyte function90 and is overexpressed on the surface of AML cells; CLEC12A could therefore serve as a target for CAR T cells to negate minimal residual disease91 or target LSC.92
TOLERANCE OF CHIMERIC ANTIGEN RECEPTOR-ENGINEERED T CELLS IN ACUTE MYELOID LEUKAEMIA
Off-Target Side Effects and Haematological Tolerance
Given the lack of specificity of the target antigens for the tumour cells, CAR T cells may have an action on normal tissues expressing these antigens. In addition, a number of these target antigens could be expressed by normal HSC and therefore myelosuppression would be expected as a consequence. However, the target selection process and the representability of the murine models made it possible to avoid major haematological toxicity in clinical trials with CAR T cells targeting CD3352 and LeY.67
Cytokine Release Syndrome
By linking target antigen-expressing malignant cells to cytotoxic T cells, T cell-engaging therapies harness the cell-mediated immune response and direct it against malignant cells, bypassing the major histocompatibility complex. Clinical trials using CAR T cells have demonstrated high complete response rates;10,26,89 however, complete remissions have been associated with CRS, the intensity of which ranges from mild to life-threatening.9,34,93 Mild CRS symptoms are characteristic of flu-like syndrome, with fevers and myalgias, while some patients experience a severe inflammatory syndrome, including vascular leak, hypotension, pulmonary oedema, and coagulopathy, resulting in multiorgan system failure. The pathophysiology of CRS is related to a significant increase in the release of cytokines, such as IL-10 and IL-6, due to the activation and proliferation of T lymphocytes.94 Among the cytokine storm, IL-6 appears to be the central mediator of CRS94 and clinical studies have shown that the use of corticosteroids or an IL-6 receptor antagonist antibody (tocilizumab) allows CRS control induced by CAR T cells.9 Other approaches, such as adding a suicide gene and kill-switch strategies,15 are possible. Due to the increased experience of corticosteroid and tocilizumab use, a consensus is emerging for the management of CRS.94 CRS does not spare AML patients treated with CAR T cells and has been reported after the use of CAR T cells against CD3352 and CD123.95
Other Toxicities
The development of neurologic toxicities, including confusion, delirium, myoclonus, aphasia, and seizure, has been reported in patients receiving CD19-specific CAR T cells.23,24 However, these side effects are also found with blinatumomab96 and seem specific to CD19, so should therefore not be found in AML. Anaphylaxis can also occur because CAR T cells contain antigen-recognition domains derived from murine antibodies97 and efforts are ongoing to humanise the components of expressed proteins. The risk of insertional oncogenesis in human cells has also been established in the context of gene therapy of HSC.98 Insertion of a transgene into T cells may induce malignant transformation; however, no cases of transformation have been reported following the infusion CAR T cells.
CONCLUSION AND REMAINING CHALLENGES
The progress of immunotherapy brings new hope for cancer treatment. Like in ALL, CAR T cells provide much hope for the treatment of AML. However, unlike in ALL, there is no target antigen such as CD19 or CD22 in AML, and the target choice can lead to off-target effects. CD123 and CD33 are the major targets, either separately or on constructions targeting both at the same time,99 and results of the numerous ongoing clinical trials need to be awaited to weigh the potential benefits versus side effects of these new strategies. Moreover, CAR T cells are currently reserved for relapsed or refractory AML patients; therefore this new strategy will have to find its place in a therapeutic arsenal that already contains alloHSCT as well as new targeted therapies. In order to reduce off-target toxicities, several strategies may be used. The identification of novel leukaemia-associated antigens or neoantigens could provide more specific targets and transcriptomic and proteomic analyses are ongoing to fully characterise the AML ‘surfaceome’. Furthermore, dual-targeting or sequential approaches could improve treatment specificity while relying on combinations of AML antigens. In addition, the understanding and prevention of the escape mechanisms of CAR T cells in AML remains a challenge. The strategy to address the relapses due to antigen escape involves designing CAR T cells able to target multiple antigens simultaneously or sequential strategies. Moreover, relapses in AML are caused by the persistence of LSC, which do not necessarily express the same antigens as the bulk cells, and therefore LSC could persist despite the use of CAR T cells. The exorbitant cost of these new strategies will also be a societal challenge, but the democratisation of CAR T cells may lead to lower costs. Therefore, CAR T cells could competitively challenge the prominent place of alloHSCT for the treatment of AML.





