Abstract
Diffuse large B cell lymphoma (DLBCL) is the most common histological subtype of non-Hodgkin’s lymphoma. However, splenic DLBCL is a relatively uncommon form of non-Hodgkin’s lymphoma.1 In this case report, the authors present a 38-year-old male who was admitted to the hospital with a complaint of abdominal distension, left-sided abdominal pain, loss of weight, and loss of appetite for 2 months. The basic workup of this patient was suggestive of microcytic anaemia with a raised total lymphocyte count, platelet count, and erythrocyte sedimentation rate, while a nodular spleen with altered splenic echotexture was revealed on ultrasonography. Splenic malignancy was suspected and contrast-enhanced CT of the abdomen was planned, which gave an impression of an extra splenic mass lesion causing impingement on the spleen with continuity to bowel loops and thickening of the fascia, raising the possibility of a gastrointestinal stromal tumour; however, the presence of large conglomerated necrosed lymph nodes in the abdominal cavity pointed the diagnosis towards a splenic lymphoma. The splenectomy specimen had multiple nodular deposits and immunohistochemistry studies finally provided a clear-cut diagnosis of DLBCL–mucosa-associated lymphoid tissue lymphoma.
Key Points
1. Splenic diffuse large B cell lymphoma is an extremely rare malignancy and has been identified in less than 1% of patient with non-Hodgkin’s lymphoma.2. It is important for the physician to consider splenic lymphoma as a differential diagnosis whenever a patient presents with nodular splenomegaly.
3. Diffuse large B cell lymphoma of the spleen is potentially curable with R-CHOP chemotherapy and splenectomy if detected in early stages of the disease.
INTRODUCTION
B cell non-Hodgkin lymphomas (NHL) are characteristically divided into aggressive and indolent tumours, out of which diffuse large B cell lymphoma (DLBCL) remains the most common and aggressively behaving NHL. DLBCL involves lymphoid tissue commonly, but non-lymphoid tissues are affected in up to 40% of patients. Due to functional heterogeneity and omnipresent distribution of lymphatic tissue, DLBCL can occur in bone marrow, central nervous system, gastrointestinal tract, liver, skin, and thyroid gland.1 However, splenic DLBCL is extremely rare and has been identified in less than 1% of patients with NHL.
Nodular splenomegaly can have many differentials including haemangioma, lymphangioma, gastrointestinal stromal tumour, abscess, infarct, metastatic deposits, and hamartoma. Ultrasonography and contrast enhanced CT (CECT) abdomen has an important role in the establishment of a specific diagnosis. DLBCL is potentially curable and responsive to combination chemotherapy with 6 cycles of R-CHOP (Rituximab, Cyclophosphamide, Hydroxydaunorubicin, Vincristine, Prednisone) every 3 weeks.
Here, the authors present a case of splenic DLBCL–mucosa-associated lymphoid tissue (MALT) lymphoma in a 38-year-old male who visited the hospital with a massive nodular spleen.
CASE REPORT
A 38-year-old male came to the emergency department with complaints of loss of appetite and loss of weight for 2 months, as well as abdominal distension, abdomen pain, and vomiting for 15 days. The patient had 3–4 episodes of vomiting per day for 15 days along with passage of dark stools. However, there was no complaint of fever, cough, breathlessness, epigastric pain, bloating, or night sweats.
On examination, the patient had pallor and dry skin. On abdominal palpation, a nodular and firm spleen approximately 11 cm from the left costal margin was detected. He had no palpable cervical, axillary, or inguinal lymph nodes. The complete blood count of this patient was suggestive of microcytic hypochromic anaemia (Hb: 6.8 mg/dL) and mean corpuscular volume, mean corpuscular haemoglobin, and mean corpuscular haemoglobin concentration values were well below the normal range. He had a raised total lymphocyte count (12.6 cmm), platelet count (493 cmm), and erythrocyte sedimentation rate (ESR; 100 mm/hr); whereas, liver functions, kidney functions, and serum electrolytes were within normal limits. Urine routine/microscopy didn’t reveal any abnormality, while Tridot, Rk-39, and rapid malarial antigen tests were also negative for this patient. However, his lactate dehydrogenase (LDH) (569 U/L) and prothrombin time–international normalised ratio (18.1/1.44) were significantly elevated.
The patient also underwent bone marrow biopsy, which revealed marked erythroid hyperplasia with dimorphic maturation and eosinophilia. Ultrasonography of the whole abdomen was done and was indicative of hepatosplenomegaly, cholelithiasis, altered splenic echotexture, and moderate ascites. The patient was finally subjected to CECT whole abdomen for a more conclusive diagnosis and imaging findings were suggestive of an extra splenic mass lesion in the left hypochondriac region causing impingement on the spleen on the medial aspect with continuity to bowel loops and thickening of fascias (possibility of gastrointestinal stromal tumour). Large conglomerated lymph nodes in the abdominal cavity with necrosis were seen. Other findings on CT revealed mild hepatomegaly, mild ascites, and cholelithiasis. The patient was immediately transferred from medicine to the surgery department as exploratory laparotomy with splenectomy was planned by the head surgeon.
Intraoperative findings post laparotomy included a grossly enlarged spleen with adhesion to the omentum and large bowel. The spleen was studded with multiple nodular deposits along with some deposits on the omentum. Due to extension of the mass to the jejunum and left colon, resection of the jejunum was done along with left hemicolectomy, omentectomy, and splenectomy. Apart from the abovementioned procedures, feeding jejunostomy and Hartmann’s procedure were also done.
All four samples of spleen, omentum, jejunum, and colon were sent for histopathology (Figure 1,2,3). Immunohistochemistry of these tumour cells was positive for CD20 and CD45 and negative for CD15, CD30, CK, and Vimentin. A final impression of MALT lymphoma-DLBCL type was made through this histopathological study.
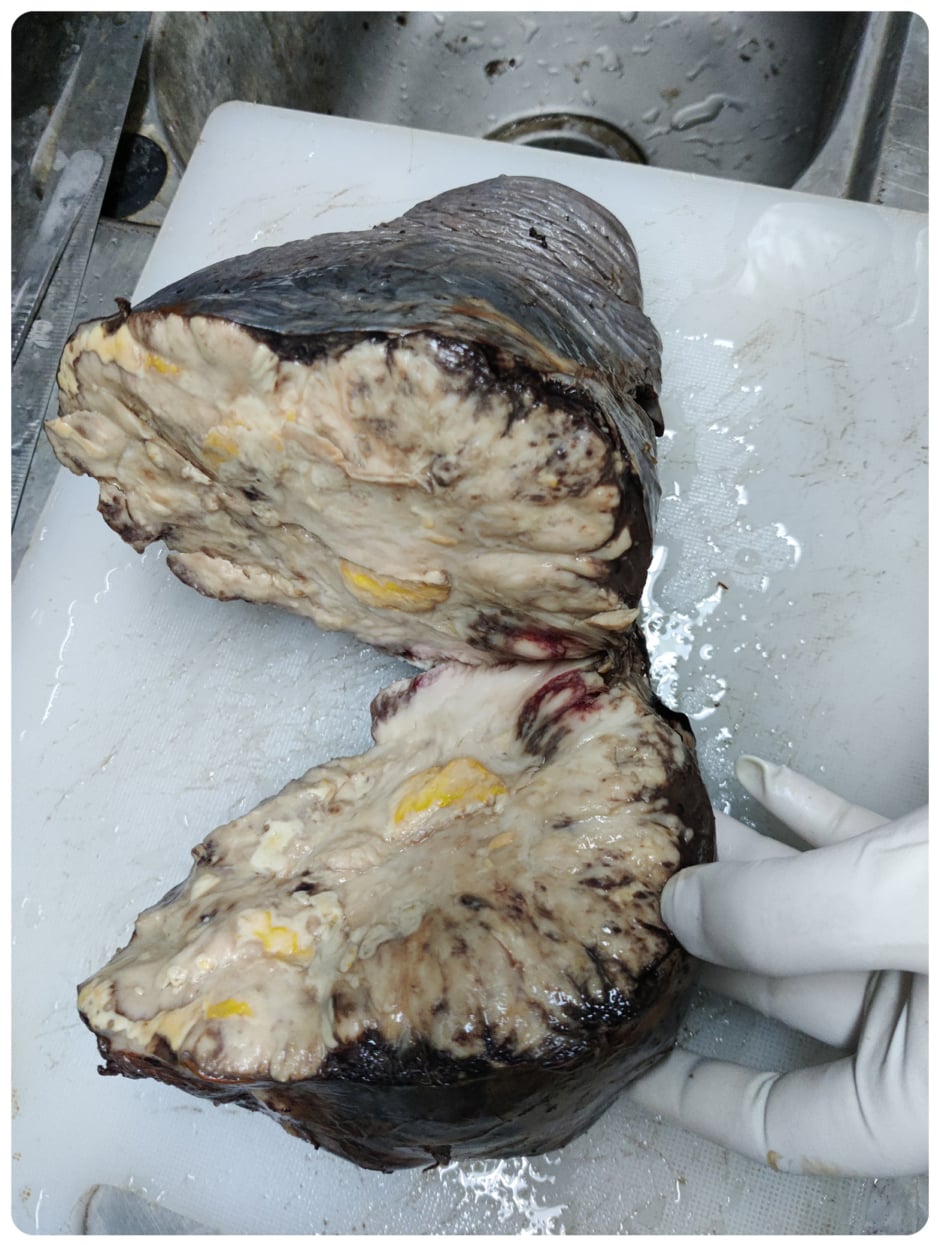
Figure 1: Gross specimen of spleen with parenchymal damage.
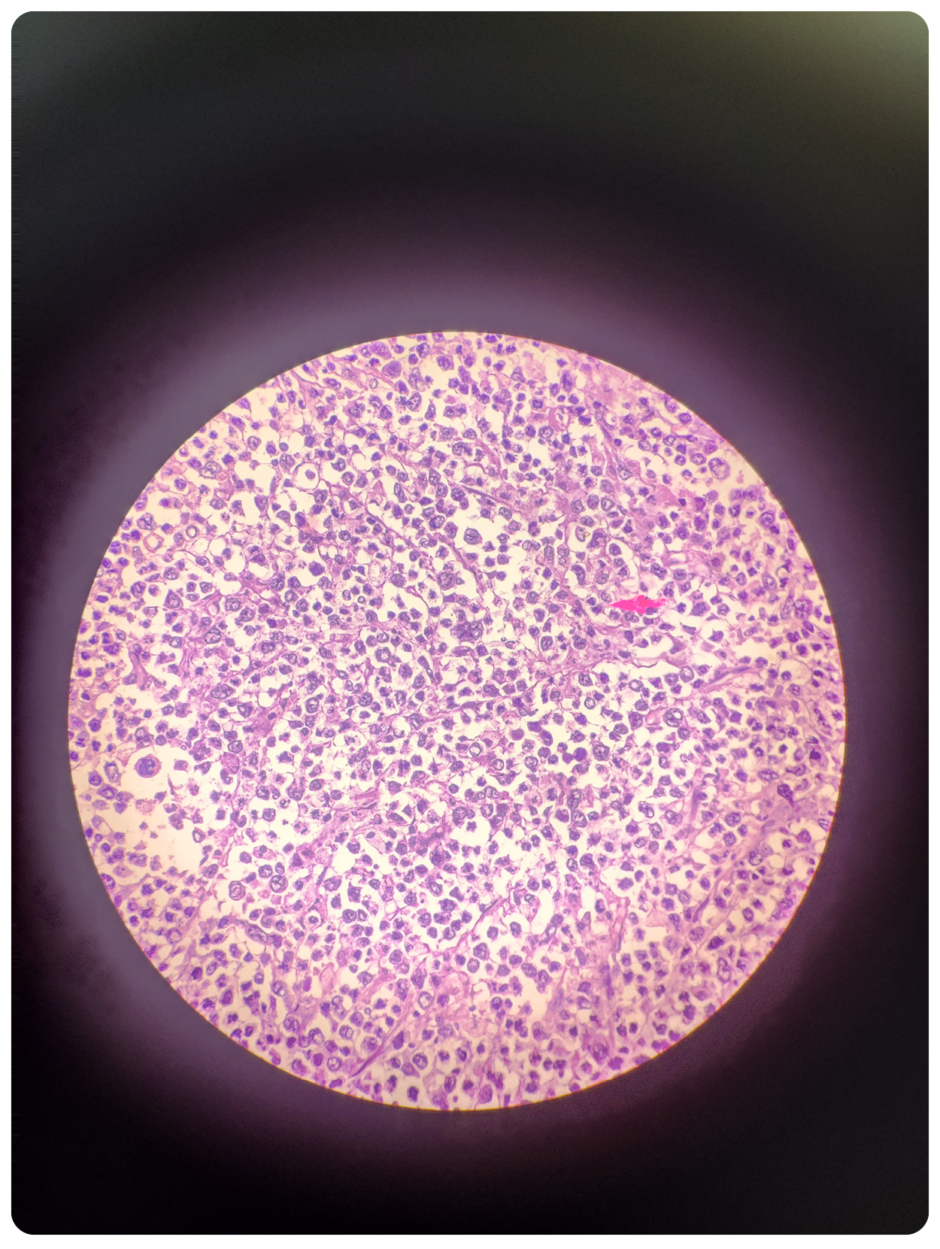
Figure 2: Microscopy of spleen: tumour cells contain large vesicular to hyperchromatic nuclei.

Figure 3: Omentum with multiple nodules consisting of tumour cells.
DISCUSSION
Of all lymphomas, 90% belong to B cell origin. DLBCL is the most common subtype of non-Hodgkin’s lymphoma. However, splenic DLBCL is a very rare type of NHL and has been detected in only <1% of patients with DLBCL. Multiple extra-nodal involvement of DLBCL indicates adverse clinical outcomes and is influenced by several oncogenic mutations as well as tumour microenvironment alterations.2 Primary splenic lymphoma (PSL) was first described by Dasgupta et al.3 as lymphoma involving the spleen and splenic hilar lymph nodes with no evidence of relapse within the first 6 months post-splenectomy. Kehoe et al.4 defined PSL as NHL arising in the spleen or restricted to the spleen and its local lymph nodes. Kraemer et al.5 approved PSL in patients with splenomegaly, cytopenia of at least two haematological cell lineages, and absence of peripheral lymphadenopathy.
DLBCL is frequently associated with infections (hepatitis C virus,6,7 hepatitis B,8 HIV, and Epstein–Barr virus), autoimmune diseases (Sjogren’s syndrome, systemic lupus erythematosus, rheumatoid arthritis) and immunosuppressed conditions. However, this patient had a negative hepatitis B, hepatitis C, and HIV status, and had no evidence of consumption of immunosuppressant drugs or any autoimmune disease. The patient with splenic lymphoma presents with symptoms of low-grade fever, abdominal distension, abdominal pain, loss of weight, loss of appetite, and night sweats.9,10 Common laboratory findings observed in patients with PSL include raised erythrocyte sedimentation rate, LDH, serum β-2 microglobulin, and cytopenia of at least two haematopoietic cell lineages. This patient had a raised erythrocyte sedimentation rate and LDH along with microcytic hypochromic anaemia, elevated TLC, and platelet count.
CECT abdomen is the preferred investigation for accurate staging of indolent lymphomas; whereas, fludeoxyglucose F18 PET is essential for diagnosing aggressive lymphomas such as DLBCL.11 Sometimes, imaging findings can be easily confused with a splenic abscess owing to the feature of liquefactive necrosis.12 The common differential diagnosis of nodular splenomegaly includes haemangioma, lymphangioma, hamartoma, abscess, metastatic deposits, or infarcts.13,14 Splenic lymphomas are usually of B cell origin, and other subtypes of NHL should be considered apart from DLBCL. Follicular lymphoma is the second leading subtype of NHL, which is characterised by small lymphocytes with cleaved nuclei and a variable population of large cells with vesicular chromatin and prominent nucleoli. Immunochemistry for follicular lymphoma is positive for Bcl2 and CD10. Follicular lymphoma may transform into diffuse large B cell lymphoma in the spleen.15 The other common NHL include marginal zone lymphomas, mantle cell lymphoma, lymphoplasmacytic lymphoma, and Burkitt’s lymphoma (the rarest NHL and most common in childhood). All NHLs are staged according to Ann Arbor staging, which focuses on the number of tumour sites (nodal and extra-nodal) and the presence or absence of B symptoms. Splenic lymphomas are categorised into three stages, Stage 1: tumours confined within the spleen only; Stage 2: involvement of the spleen along with splenic hilar lymph nodes; and Stage 3: involvement of extra splenic lymph nodes or liver.16 The abovementioned case fits into Category 3 of splenic lymphoma. DLBCL has a higher incidence among Caucasians and men aged 60 years and above.17 The median age for diagnosis of DLBCL is 64 years. Patients with splenic lymphoma do not have any evidence of lymphadenopathy and present with a normal peripheral blood smear. The tumour consists of predominantly large neoplastic cells with ample cytoplasm, vesicular chromatin, and prominent nucleoli. These cells express B cell antigens CD19, CD20 and CD79A. CD10 and BCL6 expression is suggestive of germinal centre origin whereas MUM 1 expression corresponds to non-germinal centre. Combination chemotherapy with R-CHOP (rituximab, cyclophosphamide, hydroxydaunorubicin, vincristine, and prednisone) is the standard and a potential curative therapy for DLBCL.18,19 Early stages of this disease respond to either full dose chemotherapy of R-CHOP every 3 weeks for 6 cycles or abbreviated chemotherapy for 3–4 cycles followed by involving field radiotherapy. For advanced disease DLBCL, a full course of chemotherapy is the only available option. For chemo-refractory malignancies, chimeric antigen receptor T cells are a potential curative therapy. Epigenetic mechanisms such as DNA hypermethylation or histone deacetylation normally silence gene expression. Mutations of histone-modifying genes, especially EZH2, is commonly observed in germinal centre-derived lymphomas such as follicular lymphoma and DLBCL. EZH2 is essential to the formation of germinal centre in the secondary lymphoid tissue (lymph nodes and spleen). Recently, an orally active inhibitor of EZH2, tazemetostat, has received regulatory approval for patients carrying the EZH2 mutation.20 Splenectomy with multidrug chemotherapy offers a good survival rate among patients with splenic DLBCL-MALT lymphoma.21
CONCLUSION
Splenic DLBCL-MALT lymphoma is a pretty rare malignancy. It becomes very important for the physician to consider splenic lymphoma as a differential diagnosis. CECT abdomen can help distinguish between a splenic abscess and a malignant lesion because they look similar. Splenic DLBCL is potentially curable if R-CHOP chemotherapy is initiated in early stages of the disease. Early diagnosis with CECT abdomen, splenic biopsy, and immunochemistry can be of great help in initiating improving the survival rate of splenic diffuse large B cell lymphomas.