
Abstract
Sickle cell disease is a genetic disorder caused by sickle haemoglobin. In many forms of the disease, the red blood cells can change shape upon deoxygenation due to abnormal sickle haemoglobin polymerisation. The haemoglobin proteins stick to each other, causing the cell to have a rigid surface and sickle shape and in the process damaging the red blood cell membrane, causing the cells to become stuck in blood vessels. This deprives the downstream tissues of oxygen and causes ischaemia and infarction (which may cause organ damage), such as stroke. Incidences of the disease are found most commonly in people of African descent and less commonly in people of Mediterranean, Latino, East Indian, and Arab descent (in that order). In African countries such as Nigeria, Gabon, Ghana, and the Republic of Congo, the prevalence of the sickle cell trait is between 20% and 30%, with the disease affecting ˜2–3% of the population. Herbal formulations prepared from plants are known as phytomedicines and are effective in keeping the patient out of a crisis state and enabling them to live stable lives in society, even though the faulty S gene is not eradicated but instead managed. This review highlights some of the therapeutic options in use in the management of sickle cell disease with a view to inspiring future research on this subject.
OCCURRENCE AND PREVALENCE OF SICKLE CELL DISEASE
Sickle cell anaemia is a genetically inherited disease in which the ‘SS’ homozygous individual possesses an abnormal β-globin gene (Figure 1). A single base substitution in the gene encoding the human β-globin subunit results in replacement of β6 glutamic acid by valine, leading to the various clinical manifestations of sickle cell disease (SCD). This substitution causes a drastic reduction in the solubility of sickle cell haemoglobin (HbS) when deoxygenated. Under these conditions, the HbS molecules polymerise to form a long crystalline intracellular mass of fibres that are responsible for the deformation of the biconcave disc shaped erythrocyte into a sickle shape.
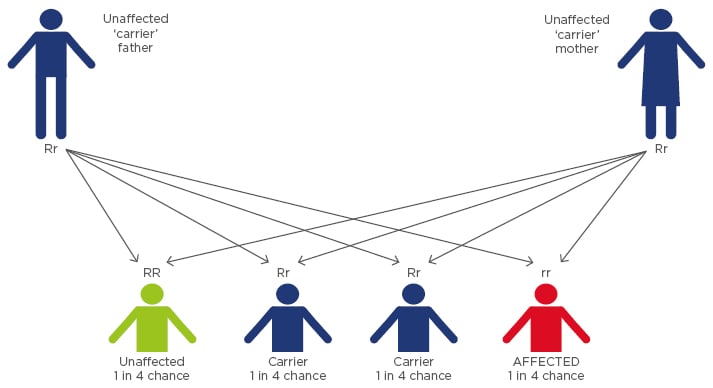
Figure 1: Sickle cell disorder inheritance pattern.
Of all the genetic disorders, SCD is the most prevalent. Incidences of the disease are found most commonly in people of African descent and less commonly in people of Mediterranean, Latino, East Indian, and Arab descent (in that order). In African countries such as Nigeria, Gabon, Ghana, and the Republic of Congo, the prevalence of the sickle cell trait is between 20% and 30%, with the disease affecting ˜2–3% of the population.1
AVAILABILITY OF TREATMENT/FIRST-LINE HEALTHCARE MANAGEMENT
First-line clinical management of sickle cell anaemia includes the use of folic and amino acid supplementation (as nutritional supplements), antibiotic/penicillin prophylaxis (to prevent infection), and anti-malarial prophylaxis (to prevent malaria attack) in varying doses in childhood, adulthood, and pregnancy. The abnormal ‘S’ gene is not eradicated in treatment, rather the condition is managed and synthesis of red blood cells induced to stabilise the patient’s Hb level. Also, transfusion therapy after screening with transcranial Doppler and vaccinations are used to manage patients.
The cost of available therapeutic options is high and not within easy reach in rural communities. Examples are blood transfusions and bone marrow transplantation. Treatments such as penicillin prophylaxis and vaccinations are very cost-effective in comparison to transfusion and hydroxyurea.
In 1984, bone marrow transplantation in a child with SCD produced the first reported cure of the disease. The transplantation was done to treat acute leukaemia, and the child’s sickle cell condition was cured as a side-event. The procedure nonetheless set the precedent for later transplantation efforts directed specifically at SCD.2
The most popular approach to prevent or reverse sickling in vitro and in vivo is to employ compounds or techniques that directly affect the Hb molecule. This could be achieved through an increase in the cell volume of erythrocytes and thus a reduction the intracellular Hb concentration below its minimum gelling concentration. This causes a delay time prior to the polymerisation of deoxygenated HbS molecules.
Another approach to affecting the Hb molecule is inducing a high concentration of fetal Hb (HbF) in sickle cell patients, which will delay/prevent HbS polymerisation. HbF differs functionally from normal adult Hb (HbA) because it has a higher affinity for oxygen. This HbF feature facilitates the binding of oxygen to Hb, giving the fetus easier access to oxygen from the mother’s blood stream.
INDUCTION OF FETAL HAEMOGLOBIN
Three types of Hb are synthesised in humans, starting from the embryonic stage (embryonic Hb, produced before birth), fetal stage (HbF, during fetal life), and from after birth (adult Hb, HbA).3 Generally, HbF usually disappears from the red blood cells of an infant soon after birth, giving way to the expression of the HbA variant of Hb. A genetic mutation at point 6 of the Beta chain of HbA gives rise to the expression of HbS, an abnormal variant.3 HbF, however, has been found to have a higher affinity for oxygen than all other variants of Hb.4 In adults, very small quantities of HbF (<2%) have been detected in the blood.3
Researchers found that SCD patients in Saudi Arabia and India that had high levels of HbF, even as adults, had milder anaemic episodes. This was due to them having inherited a genetic determinant for high HbF.5 Increasing/sustaining intracellular HBF has thus been the target of therapy aimed at an anti-sickling effect.
In the past three decades, different drugs have been produced which aimed to induce production of HbF, albeit via different mechanisms of action. Hydroxycarbamide/hydroxyurea, butyrate, 5-azacytidine, and erythropoietin are some of the drugs approved for SCD management. These drugs, together with decitabine (an analogue of 5-azacytidine) have been shown to induce HbF in vivo in animal models and sickle cell patients.2,5 Hydroxyurea was found to have the ability to increase Hb concentration and HbF values and consequently decrease the rate of pain and acute chest syndrome. Other beneficial findings associated with usage of hydroxyurea include protection against recurrent stroke and improvement in proteinuria levels. A low-dose usage of decitabine tested in a small group of SCD patients promoted a marked increase in HbF concentration and a decrease in absolute neutrophil counts. Short chain fatty acid butyrates affect chromatin structure and promote transcription rates of HbF genes. In vitro and in vivo administration of recombinant erythropoietin have been shown to increase HbF concentration when used alone and in combination with hydroxyurea.6
INCREASE IN RED BLOOD CELL VOLUME: HYDRATION
The concentration of HbS is directly correlated to cellular hydration status. Dehydrated red blood corpuscles have high intracellular HbS concentration, and as such, get to polymerise quickly. Increasing the red blood cell volume will decrease the concentration of intracellular HbS below its gelling concentration. Three major pathways are implicated in the dehydration of red blood cells: the Na+ pump, the calcium-activated potassium efflux pump, and the KCl co-transporter. Inhibition of these pathways has been shown to maintain/increase cell volume.6 Increased intracellular Mg2+ levels inhibit K+ efflux from the sickled erythrocyte and consequently prevents red blood cell dehydration. The inhibition of erythrocyte membrane ATPases with anti-sickling and anaesthetic substances and ionophoric antibiotics has been studied in the light of the partition coefficient of these drugs in erythrocyte membranes, the changes they induce in the permeability properties of erythrocytes, and their potential interaction with specific membrane components. In general, the drugs were found to inhibit both types of enzymic activity, but with varying degrees of efficacy. The (Ca2+-Mg2+)-ATPase was more sensitive to the lipophilic anaesthetics and the (Na+-K+)-ATPase to the ionophoric antibiotic, amphotericin B.
HERBAL FORMULATIONS AND PHYTOMEDICINES
Medicinal plants (also known as phytomedicines) are parts of a plant or the whole plant that possess healing properties. Folk medicine reportedly uses Carica papaya L. (Caricaceae) and Parquetina nigrescens L. (Asclepiadaceae) as a herbal remedy for the management of sickle cell anaemia.
Research into the anti-sickling properties of medicinal plants has been rewarding. This alternative therapy using phytomedicines is used in folk medicine to reduce crisis manifestations in SCD individuals. Jobelyn® (Sorghum bicolor), Ciklavit® (Cajanus cajan), Dioscovite, and Carica papaya leaf extract are among the herbal remedies used in some African countries for the management of this disease. In vitro administration of Ciklavit showed a reversal of sickling effect when analysed. Ciklavit was reported to be more potent than hydroxybenzoic acid in laboratory experiments, which revealed that the anti-sickling effect of Ciklavit may not be through nitric oxide generation or arginase inhibition, but through the induction of HbF production.7 The extract of the seeds of Cajanus cajan (a major constituent of Ciklavit) has also been found to possess anti-sickling activity. Phytochemicals responsible for this effect were found to be free amino acids, phenolic compounds (p-hydroxybenzoic acid), tannins, globulins, and saponins. Ciklavit was reported in laboratory experiments to have an anti-sickling effect through the induction of HbF production.
Anti-sickling and membrane stabilising effects of Carica papaya leaf extracts were investigated and reports indicate that the pre-treatment of sickle cell suspensions with the extract inhibited the formation of sickled cells under severe hypoxic conditions.8 Zanthoxylum zanthoxyloides (otherwise called Fagara, orin-ata) roots have also been analysed for antiprotease and membrane stabilising activity.9
Niprisan® (Nix-0699) is a herbal formulation comprising extracts of Piper guineense, Pterocarpus osun, Eugenia caryophyllum, and Sorghum bicolor. The herbal formulation reduced pain during crisis in paediatric and adult sickle cell patients. In vivo studies of Niprisan in transgenic mice showed an increase in hydrated cell volume, thus a reduction in intracellular HbS concentration and an increase in the delay time to polymerisation.10-12
Ajawaron herbal formulation (also known as Ajawaron HF) has, as its main constituent, Cissus populnea. It showed high anti-sickling activity in comparison with some controls (p-hydrobenzoic acid and n-saline). Its major phytoconstituents were found to be anthraquinone derivatives, steroidal glycosides, and cardiac glycosides.13
Earlier reports of the anti-sickling constituents of Cajanus cajan suggested cajaminose,14 phenylalanine, and hydroxybenzoic acid were responsible.15 Phytochemical studies on the aqueous extract confirmed the presence of phenylalanine and several other amino acids and phenolic compounds and tannins. The anti-sickling properties of amino acids in in vitro studies have been recognised much earlier. Of all the amino acids reported, L-phenylalanine, which was found to have anti-gelling effects, was shown to be most active.16
AMINO ACID SUPPLEMENTATION
In our daily diets, there are food substances that are rich in antioxidants and other nutritional components that help boost the immune system. Sickle cell anaemia is a genetic disease and so dietary supplements cannot stop the manifestation of the disorder, but a well-nourished and supplemented sickle cell individual can be spared the severity of the disease and go about life without the crisis episodes inherent in the disorder.
There are several compounds, such as amino acids, which prevent sickling by affecting the erythrocyte membrane, causing an increase in the cell volume of the erythrocyte and thus reducing the intracellular Hb concentration below its minimum gelling concentration. Anti-sickling properties of amino acids have been recognised much earlier; of all the amino acids reported, phenylalanine was shown to be the most active. L-phenylalanine benzyl ester (Phe-Bz), an aromatic compound, was found to be an effective anti-sickling agent at a low concentration and is therefore a potential therapeutic agent for the treatment of SCD.
ANTIOXIDANT THERAPY
A potential nutritional approach for the molecular disease found that from both in vitro and pilot clinical trials, a ‘cocktail’ of aged garlic extract, vitamin C, and vitamin E proved beneficial to patients. Ascorbic acid is important because significant oxidative stress occurs in the disease and its role as an antioxidant is very beneficial. Multivitamin supplements and a proper dietary, calorie, and protein intake are other ways to boost the immune system and extend the life span of the sickle cell individual. A scope of work on anti-sickling agents with the focus on nutrition found that the concentrations of ascorbic acid and alpha-tocopherol were significantly depressed, while that of retinol was slightly reduced in subjects tested. The depletion in the levels of the antioxidant vitamins A, C, and E may account for some of the observed manifestations of sickle cell anaemia, such as increased susceptibility to infection and haemolysis. Vitamin B12 levels have been observed to be diminished in patients with severe SCD. Patients with low vitamin B12 achieved a significant symptomatic improvement when treated with vitamin B12, 1 mg intramuscularly weekly for 12 weeks. It was concluded that many patients with severe SCD may suffer from unrecognised vitamin B12 deficiency.8
Antioxidants (scavengers of free radicals) are believed to be major components of these anti-sickling agents that add to their potential. Thus, it is believed that the higher the antioxidant property of an anti-sickling agent, the higher its potential anti-sickling effect, as this enables it to reduce oxidative stress that contributes to the sickle cell crisis. Antioxidants are found mostly in fruits (antioxidant vitamins) and vegetables. Diets rich in vitamins A, C, and E, and selenium and zinc will go a long way towards fortifying the SCD individual and preventing crisis. Research on antioxidant status and susceptibility of sickled erythrocytes to oxidative and osmotic stress has been reported using a range of diluted saline phosphate buffer in a typical osmotic fragility test to determine osmotic stress/membrane integrity and a peroxyl radical generator to induce haemolysis with oxygenated and deoxygenated red blood cells for oxidative stress analysis. It was discovered that although there are differences in antioxidant status between sickled and normal cells; these differences did not appear to be responsible for the observed difference in susceptibility to oxidative or osmotic stress-induced haemolysis.17
GENE THERAPY
Developing therapies based on the genetic manipulation of haematopoietic stem cells has long been proposed as a potential cure for sickle cell anaemia. The hallmark of a successful gene therapy includes safe and efficient gene transfer and a highly regulated and stable gene expression. A recent report on successful gene therapy in France has been widely acknowledged as a leap into the desired future of a cure for SCD. Researchers are reporting early success using gene therapy to treat, or even potentially cure, sickle cell anaemia.18 The findings come from just one patient, a teenage boy in France. The boy, now 15 years old, was treated at Necker Children’s Hospital, Paris, France, in October 2014. Researchers gave him a gene, taken up by his blood stem cells, to help prevent the sickling, but >15 months after receiving the treatment, he remained free of symptoms and his usual medications. Now, about half of his red blood cells have normal Hb; he has not needed a transfusion since 3 months after his treatment and is off all medicines.19,20
Much more research is required before gene therapy can become an option for sickle cell anaemia. It is not clear how long the benefits will last, the authors of the study said. Furthermore, the approach obviously has to be tested in more patients. A stem cell transplant from a blood-matched sibling is a potential cure, but fewer than one in five people have a donor like that. Pain crises are treated with blood transfusions and drugs, but they are a temporary fix. Gene therapy offers hope of a lasting one.4 Herein lies the future of permanent therapy for SCD.





