Ethical approval statement: Ethical approval for this study was provided by an institutional review board, and consent to participate has been taken from the patient’s guardian with informed verbal consent.
Abstract
Haemophagocytic lymphohistiocytosis (HLH) is a rare but potentially aggressive and life-threatening syndrome of overactive histiocytes and lymphocytes that commonly affects infants; it is also observed in children and adults of all ages. The disease is differentiated into either primary or secondary causes. Primary HLH tends to be of genetic origin, while secondary HLH results from either infection, autoimmune disorders, or malignancies. Secondary HLH is most commonly associated with viral infections in immunocompromised patients. This paper presents a case of HLH in a tertiary care hospital, associated with adult-onset Still’s disease, diagnosed on both biochemical criteria and histopathologic examination of bone marrow smear.
INTRODUCTION
The term haemophagocytic lymphohistiocytosis (HLH) was first described by Farquhar and Claireaux in 1952, who termed the disease familial haemophagocytic reticulosis and presented it as a rare familial disorder characterised by proliferation of histiocytes in solid organs and phagocytosis of blood cells.1 The pathophysiology and aetiology of HLH involve excessive inflammation and tissue destruction due to abnormal immune activation which is caused by the absence of normal downregulation of activated macrophages, natural killer (NK) cells, and cytotoxic lymphocytes. The most common infectious trigger is a viral infection, especially the Epstein–Barr virus (EBV). The worldwide incidence of HLH is reported to be 1.2 cases per million persons per year.
CASE PRESENTATION
A 29-year-old Asian male with no known comorbidities presented with complaints of fever (documented up to 104 °C), sore throat, skin rash, yellowish discolouration of eyes and sclera, and generalised abdominal pain for 1 month. The patient had symptoms of generalised weakness, decreased appetite, and clinically significant weight loss. He denied loose motions, easy bruising, blood in vomitus, blood in the stool, pale stools, dark urine, transfusion history, itching, a history of jaundice in his family, intravenous drug abuse, or any recent history of travelling. He was started empirically with intravenous ciprofloxacin, intravenous hydration with 0.9% normal saline, and ibuprofen for his fever, jaundice, and generalised abdominal pain.
The physical examination was unremarkable except for severe pallor; jaundice; two subcutaneous, fluctuant, hyperpigmented nodules to the right of the umbilicus; hepatomegaly; splenomegaly; and cervico-axillary lymphadenopathy. The laboratory findings revealed a haemoglobin of 6.60 g/dL, mean cell volume of 74 fL, total leucocyte count of 18.1×109 cells/L, and platelets of 57.0×109 /L. The patient had an international normalised ratio of 2.28, serum total bilirubin of 6.23 mg/dL with direct bilirubin of 5.40 mg/dL, alanine aminotransferase of 479 U/L, aspartate aminotransferase of 247 U/L, alkaline phosphatase of 799 U/L, and g-glutamyl transferase of 235 U/L. The differential considerations at this point included acute hepatitis, autoimmune hepatitis, adult-onset Still’s disease, polyarteritis nodosa, and cytokine storm secondary to COVID-19.
Further workup showed negative viral serologies, ferritin of 40,000 ng/mL, erythrocyte sedimentation rate of 45 mm/hour, C-reactive protein of 93 mg/L, serum lactate dehydrogenase of 1470 U/L, fibrinogen of 0.57 g/L, serum albumin of 3.26 g/dL, and triglycerides of 234 mg/dL. The peripheral smear showed microcytic, hypochromic anaemia with positive pencil cells and right-sided neutrophil shift. Procalcitonin was within a normal range and blood culture was negative. An ultrasound of his abdomen showed hepato- and splenomegaly (17 cm and 19 cm respectively). All other investigations were unremarkable. An autoimmune profile was negative with normal complement levels. The patient fulfilled Yamaguchi criteria of adult-onset Still’s disease,2 including high spiking fever, evanescent rash, sore throat, hepatosplenomegaly, leukocytosis, elevated polymorphonuclear neutrophil count, high erythrocyte sedimentation rate, high serum ferritin, and elevated liver enzymes, with negative tests for antinuclear antibody and rheumatoid factor (IgM). Confirmatory tests for Yersinia enterocolitica and Mycoplasma pneumoniae infections were negative, which are considered possible causes of Still’s disease. CT scan with contrast showed mildly prominent abdominal wall vessels suggestive of vasculitis, along with axillary and cervical lymphadenopathy and splenomegaly (Figure 1). Histology of the hyperpigmented abdominal nodules following punch biopsy showed leukocytoclastic vasculitis.
Bone marrow aspirate was performed which showed macrophages with classic haemophagocytic activity suggestive of HLH (Figure 2). According to the HLH-2004 protocol,3 this patient fulfilled six features out of the eight-point diagnostic criteria for HLH, including a persistent fever, bicytopenia, hepatosplenomegaly on an ultrasound of the abdomen, low fibrinogen levels, raised ferritin levels, elevated triglycerides, and histiocytic activity on a bone marrow aspirate (Table 1). In this case, the underlying cause of HLH was suspected to be adult-onset Still’s disease. The patient was given high-dose corticosteroids as a pulse therapy (750 mg methylprednisolone intravenously once daily (qd) for three days) and cyclosporine, and was planned for allogeneic bone marrow transplantation. In the course of their hospital stay, the patient’s condition deteriorated and after 7 days in the intensive care unit he could not be revived following cardiopulmonary arrest secondary to sepsis and multiple organ failure.
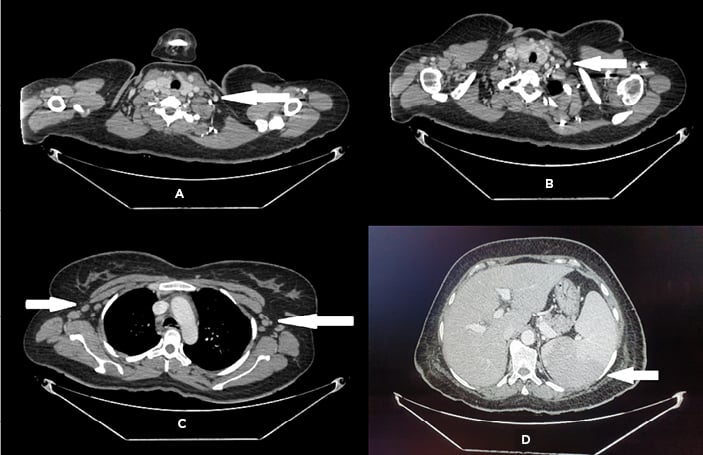
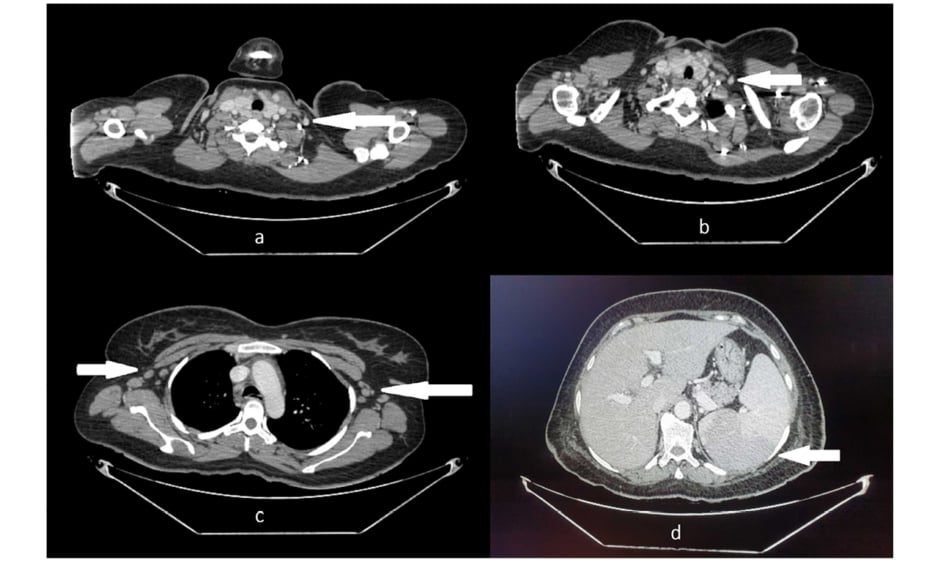
Figure 1: CT of A,B) neck; C) chest; and D) abdomen, showing bilateral cervical and axillary lymphadenopathy and splenomegaly.
The images were provided by Dow University Hospital’s medical record with the permission of the relevant head of department. Written consent was obtained from the patient’s guardian to include the images for the purpose of medical publication.
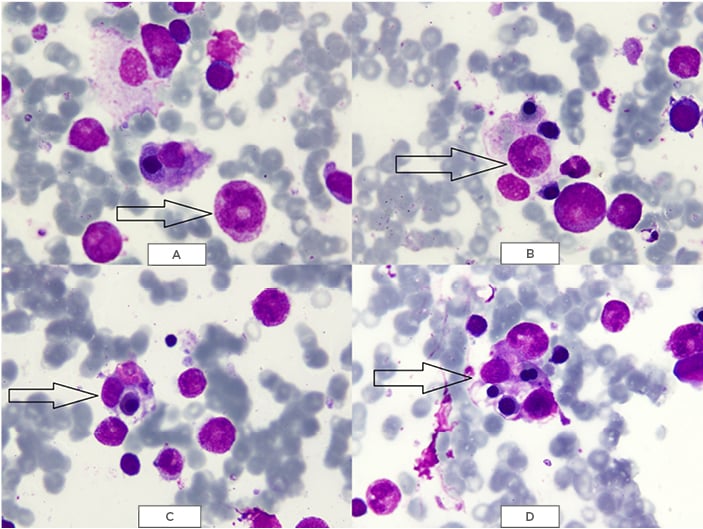
Figure 2: May–Grünwald–Giemsa (MGG) staining of bone marrow showing A,B) phagocytic cells containing erythrocytes and platelets; and C,D) macrophages phagocytising erythrocytes and platelets.
The images were provided by Dow University Hospital’s medical record with the permission of the relevant head of department. Written consent was obtained from the patient’s guardian to include the images for the purpose of medical publication.
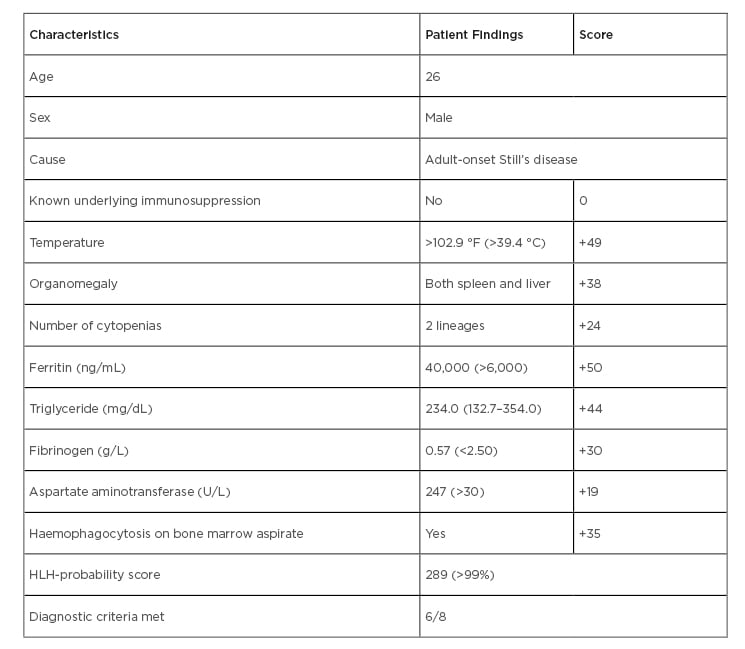
Table 1: General characteristics and HLH-probability scores of the patient.
DISCUSSION
HLH is a rare but aggressive and life-threatening syndrome of overactive histiocytes and lymphocytes that commonly affects infants, but is also observed in children and adults of all ages. Primary HLH, also known as familial haemophagocytic lymphohistiocytosis (FHL), is an autosomal recessive disease caused by a gene mutation of the FHL loci.4 The incidence of FHL is 0.12 out of 100,000 births annually, with a male-to-female ratio of 1:1,5 whereas in adults there is a slight male predominance.6 Secondary HLH, also known as sporadic or acquired HLH, describes patients without a known familial mutation and who typically have a clear trigger for acute HLH development, such as viral infection, autoimmune disease, immunodeficiency, or underlying malignancy. Macrophage activation syndrome is a form of HLH that occurs primarily in patients with rheumatologic diseases, commonly systemic-onset juvenile idiopathic arthritis, adult-onset Still’s disease, and systemic lupus erythematosus.7
The pathophysiology and aetiology of HLH involve excessive inflammation and tissue destruction due to abnormal immune activation which is caused by the absence of normal downregulation of activated macrophages, NK cells, and cytotoxic lymphocytes.8 In patients with genetic predisposition, and in sporadic cases with no underlying genetic cause identified, immune activation from an infection is a common trigger. The most common infectious trigger is a viral infection, especially EBV.9 The worldwide incidence of HLH is reported to be 1.2 cases per million persons per year. This amounts to 1 case per 50,000 births. The age of onset is usually younger than 1 year for the familial form and after 6 years for the secondary sporadic form, but adult-onset has also been reported.10-12
The presentation of HLH is of a febrile illness associated with multiple organ involvement, presenting as malaise, hepatosplenomegaly, jaundice, generalised lymphadenopathy, and cytopenias.3 Central nervous system symptoms include seizures, meningitis, encephalopathy, ataxia, hemiplegia, cranial nerve palsies, mental status changes, or irritability seen in up to 75% of paediatric cases.12 The diagnostic criteria of HLH, introduced by the Histiocyte Society (HS) in 1991,13 include fever, splenomegaly, cytopenia affecting at least two out of three lineages, hypertriglyceridaemia and/or hypofibrinogenaemia, and haemophagocytosis in the bone marrow, spleen, or lymph nodes. These criteria were updated in 20043 to include low or absent NK cell activity measured by the chromium-51 release assay, elevated soluble interleukin-2 receptor a (IL-2Rα/sCD25), and elevated ferritin. Of these eight criteria, at least five are required for diagnosis, although in patients with FHL mutations the diagnosis can be made without meeting five criteria.14
The histopathologic findings of HLH include the accumulation of lymphocytes and mature macrophages, which occasionally exhibit haemophagocytic activity, typically seen in the bone marrow but can also occur in the spleen, lymph nodes, liver, skin, lungs, meninges, CSF, and, rarely, subcutaneous tissue.15 A definitive diagnosis of HLH can be made by a molecular analysis that includes a demonstration of PRF1 mutation based on flow cytometry by staining perforin contained in lymphocytes.16 Mutation analysis should be performed in all HLH cases because the demonstration of a characteristic genetic defect alone can be used to diagnose HLH without five of the eight diagnostic criteria being met.3,11-14
A large number of underlying conditions have been reported in association with HLH.17 The management of HLH aims to suppress the exaggerated immune response using immunosuppressive agents. The second international meeting of the HS in 2004 formulated a treatment protocol for HLH which recommends an 8-week induction therapy with corticosteroids, etoposide, and cyclosporine.18 Corticosteroids suppress the elevated cytokines, cyclosporine inhibits T-cell activation, and etoposide blocks cell division and proliferation. Stem cell transplant is indicated in selective familial cases as it can improve 3-year survival from nearly 0% to 50%. Some cases have reported stem cell transplantation to be curative.19 The prognosis of genetic HLH without therapy is poor, with a median survival of 1–2 months,20 and 3-year survival <10%.18 Macrophage activation syndrome in systemic-onset juvenile idiopathic arthritis has a reported mortality of 8–22%.7 The overall reported mortality for acquired HLH exceeds 50%. Among all the viruses associated with HLH, EBV carries the worst prognosis, with reported mortality ranging from 25% to 100%.9 However, the addition of etoposide improves prognosis if given within the first 4 weeks.7
In this review of the literature, data mining was performed using the PubMed search engine and Google Scholar, using medical terms involving or similar to ‘adult-onset Still’s disease associated with HLH.’ A variety of cases were found to report an association of HLH with Still’s disease. One of the cases presented with severe pancytopenia and jaundice, which are the most likely presenting features of both illnesses.21 Still’s disease usually has a good prognosis with steroids, but the therapy has been reported to cause gastrointestinal ulceration.22 Rarely, the course of both diseases can be complicated with disseminated intravascular coagulation.23 M. pneumoniae has been known to cause HLH on a background of Still’s disease.24 One review article suggested serum ferritin levels as a diagnostic marker of HLH, and a marker of disease activity for Still’s disease. However, hyperferritinaemia can be a feature of many uncommon pathologies including autoimmune, infectious, inflammatory, or neoplastic, where it plays a pro-inflammatory role which can sometimes lead to a cytokine storm.25 One study suggested the presence of interstitial lung disease is a significant risk factor for developing HLH, as well as relapsing of Still’s disease.26 Lastly, high-dose steroids have been the mainstay of treatment whenever there is an overlapping presence of Still’s disease with features of HLH.27
CONCLUSION
This case report highlights a case of HLH in association with adult-onset Still’s disease, which fulfilled five out of eight defined criteria outlined by the HS, including fever, hepatosplenomegaly, cytopenia, hypertriglyceridaemia, hypofibrinogenaemia, haemophagocytosis in the bone marrow, and elevated ferritin. Treatment focussed on the elimination of the triggering infection, and utilised high-dose glucocorticosteroids along with second-line therapies of cyclosporin, intravenous immunoglobulins, and etoposide, but the patient did not survive.





