BACKGROUND AND AIMS
The KMT2A-MLLT3 (also known as MLL-AF9) fusion is associated with acute lymphoid leukaemia (ALL), acute myeloid leukaemia (AML), and mixed phenotype acute leukaemia (MPAL) in infants and children. Infant leukaemia originates in utero, yet this has never been formerly shown for KMT2A-MLLT3-driven disease. Multiple mouse models invariably revealed that KMT2A-MLLT3 can induce myelo-monocytic AML; however, they failed to recapitulate the ALL and MPAL phenotypes as presented in paediatric patients. To explore the impact of the developmental stage for KMT2A-MLLT3+ leukaemic burden and phenotypes, the authors established a new inducible (‘Tet-off’), reversible, transplantable mouse model in which expression of the fusion is controlled by a stem cell leukaemia (SCL)-enhancer element (Figure 1).
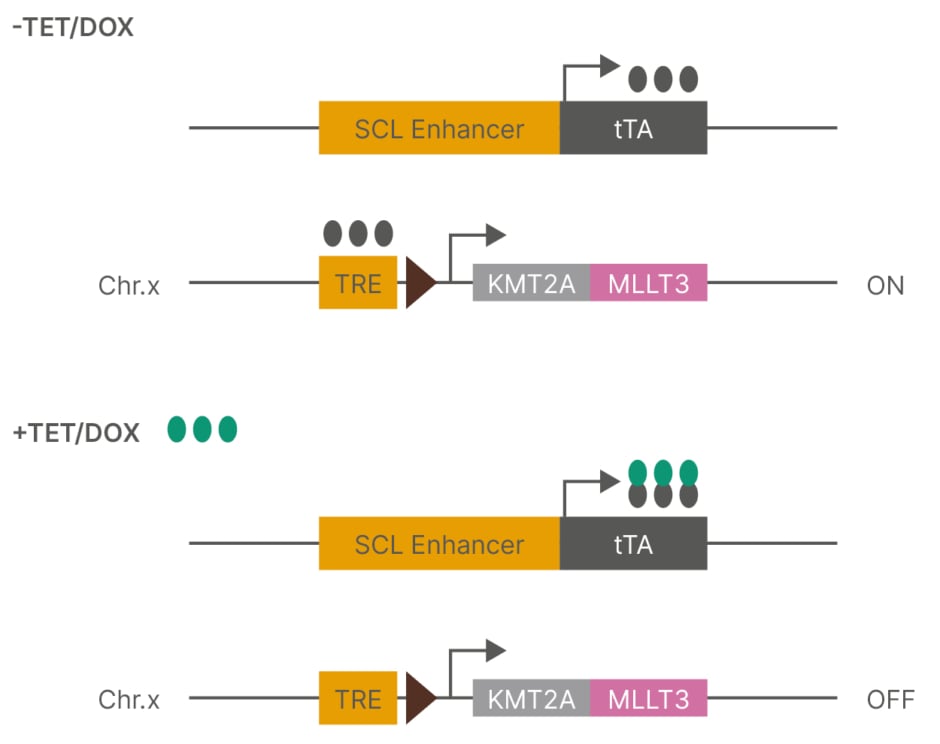
Figure 1: Tet-Off system.
Chr.x: chromosome X; DOX: doxycycline; SCL: stem cell leukaemia; TET: tetracycline; TRE: tet-responsive element; tTA: TET-controlled transactivator.
MATERIALS AND METHODS
The authors crossed SCL-tTA mice with inducible KMT2A-MLLT3 mice1 to generate double transgenic SCLtTA/i KMT2A-MLLT3 mice. To determine the impact of the developmental stage, KMT2A-MLLT3 was induced from the conceptional stage for prenatal (E0), in neonates for postnatal (P0), and at 20 weeks for adult (≥20 weeks) mouse cohorts. Following induction for 6–8 weeks in vivo KMT2A-MLLT3 expression was measured in haematopoietic stem cells (HSC), haematopoietic progenitor cell-1/lymphoid-myeloid primed progenitors (HPC-1/LMPP), and granulocyte-monocyte progenitors (GMP). Notably, the KMT2A-MLLT3 expression levels were comparable within each population across the age groups.
RESULTS
Upon postnatal KMT2A-MLLT3 induction, all the mice developed leukaemia with a short median latency of 13 weeks and complete penetrance. In contrast, E0 prenatal- or adult-induction resulted in longer latencies of 32 and 24 weeks, respectively. Despite sustained KMT2A-MLLT3 expression, not all the mice developed leukaemia in prenatal- and adult-induced cohorts. Postnatal-induced mice developed myelo-monocytic, megakaryoblastic, or MPAL, whereas prenatal and adult induction of KMT2A-MLLT3 developed myelo-monocytic disease exclusively. Regardless of leukaemia phenotypes, all diseased mice displayed splenomegaly, and only postnatal and adult sick mice developed thrombocytopenia. Independent from the age at which KMT2A-MLLT3 was induced, all myelo-monocytic AML mice had clonal expansion of GMPs. Only postnatal-induced myelo-monocytic mice had a significant increase in HPC-1/LMPPs and reduction in megakaryocyte erythrocyte progenitor and multi-potent progenitor populations, relative to age-matched controls. There was expansion of GMPs and HPC-1/LMPP populations in the postnatal-induced MPAL disease; whereas, in the postnatal-induced megakaryoblastic disease, there was clonal expansions of HSCs, LK (lineage negative c-kit positive), and MEP populations with no expansion in GMP. Thus, the age when the KMT2A-MLLT3 fusion is induced does not only determine the disease latency and penetrance, but also affects lineage determination and the amount of leukaemic HSPCs.
Leukaemic-GMP and leukaemic-HSC have been characterised in adult models as leukaemic stem cells. Indeed, transplantation of bulk and leukaemic-GMPs from adult-induced myelo-monocytic AML cells led to secondary disease with complete penetrance. In all age cohorts, bulk myelo-monocytic AML cells formed serially plateable large and compact colonies and were reversible. Collectively, leukaemic stem cells exist within a GMP-like population of adult-induced KMT2A-MLLT3 AML, they may however reside in different HSPC populations in prenatal- and postnatal-induced disease.
CONCLUSION
In summary, the authors generated a novel inducible model that recapitulates several features of infant, paediatric, and adult KMT2A-MLLT3 leukaemia, demonstrating that the ontogenic stage is a determining factor in differences in disease biology.





