Abstract
Inflammatory bowel disease, consisting of Crohn’s disease and ulcerative colitis, causes chronic gastrointestinal symptoms and can lead to morbidity and mortality if uncontrolled or untreated. However, for patients with moderate-to-severe disease, currently available therapies do not induce or maintain remission in >50% of patients. This underscores the need for additional therapies. In this review, the authors detail the novel therapies vedolizumab, tofacitinib, and ustekinumab and delve into therapies which may come onto the market within the next 10 years, including JAK-1 inhibitors (filgotinib and upadacitinib), IL-23 inhibitors (guselkumab, mirikizumab, and risankizumab), the anti-β4β7 and anti-βEβ7 integrin monoclonal antibody etrolizumab, the sphingosine-1-phosphate subtypes 1 and 5 modulator ozanimod, and mesenchymal stem cells. Further studies are required before these emerging therapies gain approval.
INTRODUCTION
Inflammatory bowel disease (IBD), consisting primarily of ulcerative colitis (UC) and Crohn’s disease (CD), are chronic diseases affecting the gastrointestinal tract. Symptoms include diarrhoea, with and without blood, and abdominal pain. UC and CD, if left untreated or uncontrolled, cause significant morbidity and mortality at rates higher than the general population.1 Furthermore, the incidence and prevalence of IBD are increasing over time around the globe.2
Standard therapies in IBD include topical 5-aminosalicylate products such as mesalamine and sulfasalazine, thiopurines such as azathioprine and 6-mercaptopurine, and topical and systemic corticosteroids. Unfortunately, topical 5-aminosalicylate products are not recommended in moderate-to-severe UC because of a lack of efficacy in this cohort.3 Thiopurines are only effective in about one-quarter of patients,4 and corticosteroids are not recommended for maintenance of remission as a result of dose-dependent short and long-term adverse effects, such as increased risk of serious infection, weight gain, elevated blood sugar levels, bone loss, and cataracts.5, 6
Novel medications for IBD include biologics such as the anti-TNF agents infliximab and adalimumab, the anti-α4β7 integrin monoclonal antibody vedolizumab, the anti-IL-12/IL-23 monoclonal antibody ustekinumab, and small molecules inhibiting JAK such as tofacitinib. However, in patients with moderate-to-severe UC and CD, these medications have an average induction of remission and maintenance of remission rates <50%,7-13underscoring the need for additional therapies for nonresponders and those who lose response to treatment. These remission rates are even lower in patients with prior exposure to biologic therapy. In this paper, the authors discuss select new IBD therapies and emerging therapies that will likely come onto the market in the next 5–10 years (Table 1).8-26
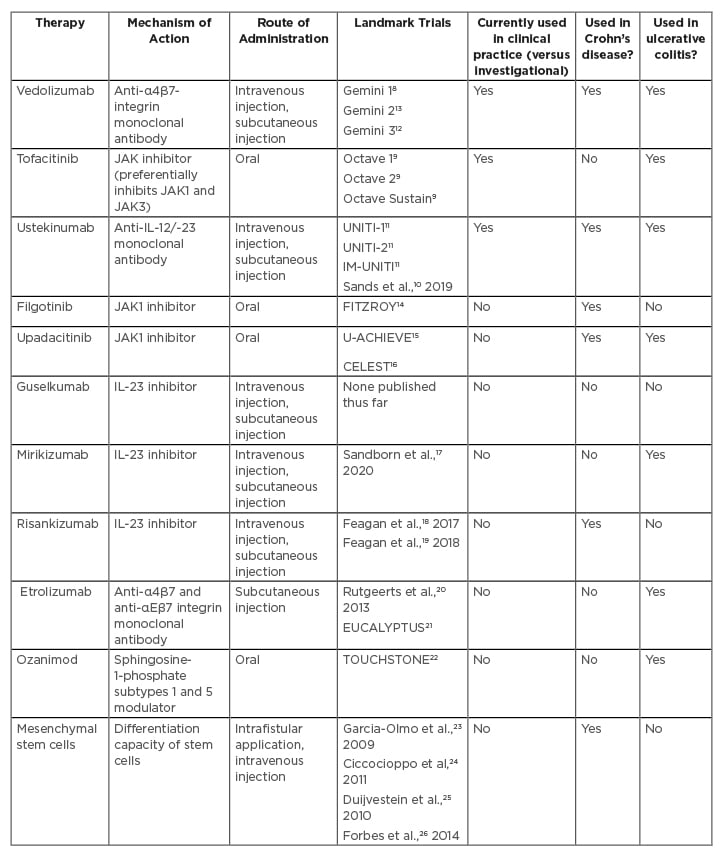
Table 1: Overview of ovel and emerging therapies in inflammatory bowel disease.
NOVEL THERAPIES
Vedolizumab
Vedolizumab is an anti-α4β7 integrin monoclonal antibody that modulates gastrointestinal tract inflammation by inhibiting adhesion of peripheral blood lymphocytes to MAdCAM-1.27 Because of this molecule’s specificity to the gastrointestinal tract, the risk of progressive multifocal leukoencephalopathy (PML) is markedly decreased compared to that of natalizumab, an anti-α-4-integrin monoclonal antibody that causes systemic immunosuppression.27
In the Gemini 1 trial, in patients with moderate-to-severe UC, patients were randomised to either vedolizumab or placebo for induction.8 In the maintenance arm, patients who responded to induction therapy were randomised to vedolizumab every 8 weeks, vedolizumab every 4 weeks, or placebo for up to 52 weeks. Higher rates of clinical response (the primary outcome) at Week 6 were seen in the vedolizumab arm than the placebo arm (47.1% versus 25.5%; p<0.001). Significantly higher rates of clinical remission were seen in the vedolizumab arms compared to placebo. Rates of serious infections were similar between the arms, including no cases of PML in either group.
The Gemini 2 and 3 trials studied vedolizumab in moderate-to-severe CD. Gemini 2 had a similar trial design as that of Gemini 1.13 More patients on vedolizumab were in remission at Week 6 compared to placebo (14.5% versus 6.8%; p=0.02). In the maintenance arm, significantly more patients on vedolizumab were in remission at Week 52 than those on placebo. Of the most common adverse events (AE), only nasopharyngitis occurred more frequently in the vedolizumab arm. No cases of PML were reported and infections, serious infections, and serious AE occurred more frequently in the vedolizumab arm.
The Gemini 3 trial focussed on patients in whom prior therapy had failed, with 76% of patients having experienced anti-TNF failure12 (whereas in Gemini 2 the number of patients who had failed these agents was limited to 50% of the total).13 A nonsignificantly increased portion of anti-TNF agent nonresponders in the vedolizumab arm were in clinical remission at Week 6 versus those on placebo (15.2% versus 12.1%; p=0.443). A significantly higher number of patients were in clinical remission at Week 10 in the vedolizumab arm compared to placebo (26.6% versus 12.1%; nominal p=0.001). Rates of serious infections and serious AE were similar between the vedolizumab and placebo arms. Gastrointestinal infections occurred in slightly more patients in the vedolizumab arm. No cases of PML were reported.
Subcutaneous vedolizumab may be preferred by many patients given its convenience as compared to intravenous administration. In a UC trial of this formulation, patients received two doses of intravenous vedolizumab.28 Those who responded were randomised to subcutaneous vedolizumab, intravenous vedolizumab, or placebo. Significantly more patients in the subcutaneous vedolizumab arm achieved clinical remission at Week 52 than in the other two arms. Safety profiles were similar between the subcutaneous and intravenous vedolizumab arms.
Results from the study investigating subcutaneous vedolizumab in CD have only been presented in abstract form.29
Tofacitinib
Tofacitinib is a small molecule that inhibits all JAK pathways implicated in the pathogenesis of IBD, especially JAK1 and JAK3.30 Small molecules have some advantages over biologics, such as their ability to be administered orally. Furthermore, small molecules do not induce a host antibody response so there is no need for concomitant administration of immunomodulators (such as azathioprine, 6-mercaptopurine, or methotrexate) that carry their own toxicities and AE. Overall, these properties suggest that small molecules are easier to administer, may have a more durable response, and may have a favourable safety profile in comparison to combination of anti-TNF and immunomodulator therapy.
Results from the Octave Induction 1, Octave Induction 2, and Octave Sustain trials demonstrated the effect of tofacitinib on patients with moderate-to severe-UC.9 In the Octave Induction 1 and 2 studies, patients were randomised to receive either tofacitinib 10 mg twice daily or placebo for 8 weeks. In the Octave Sustain trial, patients with a response to induction therapy were randomised to tofacitinib at either 5 or 10 mg twice daily or placebo for 52 weeks. In both induction trials, tofacitinib produced a significantly higher rate of clinical remission at Week 8. Similarly, maintenance of remission at 52 weeks was significantly higher in both tofacitinib groups.
Worsening of UC, nasopharyngitis, headache, and arthralgias were the most common AE reported. Serious AE were not more common in the tofacitinib arms. Infection rates were higher in the tofacitinib arms in both induction and maintenance trials. Five patients who had received tofacitinib experienced cardiovascular events. One episode of gastrointestinal perforation occurred in a patient with cytomegalovirus infection on prednisone. The discovery of increased rates of pulmonary embolism and mortality in older rheumatoid arthritis patients with at least one cardiovascular risk factor receiving higher dose tofacitinib31 led the U.S. Food and Drug Administration (FDA) to issue a black box warning about tofacitinib.31,32 Tofacitinib should be reserved for patients failing biologic therapy and it is recommended that the dose is reduced to 5 mg twice daily after successful induction with 10 mg twice daily. Results of the effect of tofacitinib in CD have not been as promising as in UC.33,34
Ustekinumab
Ustekinumab is a monoclonal antibody against the p40 subunit of the IL-12/-23 receptors. Results from the Phase III trials (UNITI-1, UNITI-2, IM-UNITI) were published after promising results from Phase IIa and IIb trials,35,36 which consisted of an 8 week induction trial and 44 week maintenance trial in patients with moderate-to-severe CD.11 The UNITI-1 trial consisted of patients who had primary nonresponse, secondary nonresponse, or AE from anti-TNF agents. The UNITI-2 trial consisted of patients who did not respond to or experience AE from immunosuppressants or glucocorticoids. In both the UNITI-1 and UNITI-2 trials, patients in either ustekinumab arm were more likely to achieve the primary endpoint (clinical remission at Week 6) compared to placebo. Similarly, in the IM-UNITI trial, more patients on ustekinumab were in clinical remission at Week 44, compared to patients on placebo. Rates of AE, including serious AE, were similar across arms in the induction and maintenance trials. Long-term extension trial results have been similarly promising.37
In an 8 week induction trial studying ustekinumab in UC, patients were assigned either fixed or weight-based doses of ustekinumab or placebo.10 Patients that responded to ustekinumab were then randomised to one of two frequencies of ustekinumab or placebo in the 44-week maintenance trial. Patients in both ustekinumab arms of the induction trial were more likely to achieve clinical remission than those given placebo (p<0.001 for both comparisons). Patients in both ustekinumab arms of the maintenance trial were significantly more likely to achieve clinical remission than those in the placebo arm. In the induction trial, AE were similar across all three arms, with serious AE highest in the placebo arm. In the maintenance trial, higher rates of AE, including serious AE, were reported in the placebo arm.
EMERGING THERAPIES
JAK1 Inhibitors
Filgotinib
Whereas tofacitinib inhibits all JAK with a preference for JAK1 and JAK3,30 filgotinib selectively inhibits JAK1 only.38 In the Phase II FITZROY study, the safety and efficacy of filgotinib was tested in patients with moderate-to-severe CD.14 In the first 10 weeks, patients were randomised to either filgotinib or placebo. In the following 10 weeks, patients were stratified based on prior clinical response, prior anti-TNF agent exposure, and baseline corticosteroid use, among other considerations, to one of two doses of filgotinib or placebo. More patients in the filgotinib group achieved clinical remission than those in the placebo group in the induction phase of the trial (47% versus 23%; p=0.0077). AE were similar between treatment and placebo arms, but serious AE occurred at higher rates in the treatment arm, including four patients who developed serious infections. Additional trials studying filgotinib are underway in CD39,40 and UC.41,42
Upadacitinib
Upadacitinib is a JAK inhibitor that selectively binds JAK1,43 similar to filgotinib. This small molecule has been studied in Phase II trials in both UC and CD.15,16 Results from the induction trial of the Phase IIb trial U-ACHIEVE, studied upadacitinib in moderate-to-severe UC.15 All patients in the study had inadequate response to or loss of response to corticosteroids, immunosuppressives, or biologics. The study consisted of two 8-week parts. In the first part, eligible patients were randomly assigned to receive one of four doses of oral upadacitinib or placebo. In the second part, eligible patients were randomly assigned to one of two doses of upadacitinib.
The primary endpoint of clinical remission at 8 weeks was achieved in more patients receiving upadacitinib than those receiving placebo. Of the patients receiving the 7.5 mg dose, 8.5% achieved the primary endpoint. This increased to 14.3% in those receiving the 15 mg dose, 13.5% receiving the 30 mg dose, and 19.6% receiving the 45 mg dose, whereas 0.0% achieved clinical remission with placebo (respective p value comparisons with placebo: p=0.052, p=0.13, p=0.011, and p=0.002, respectively). Fewer patients who had previously failed anti-TNF agents responded to upadacitinib. More patients on upadacitinib demonstrated biologic, endoscopic, and histologic response. Rates of AE were similar between patients receiving upadacitinib and placebo, including serious AE and serious infections. Further studies of upadacitinib in UC are underway.44,45
Upadacitinib was studied in moderate-to-severe CD in the Phase II CELEST trial.16 In the 16-week induction trial, patients were randomised to one of five upadacitinib doses or placebo, stratified by prior anti-TNF use and endoscopic disease severity. After the induction trial, patients were rerandomised to one of two upadacitinib doses or placebo for the 36-week maintenance trial. In the induction trial, the proportion of patients achieving clinical remission, the primary endpoint, or endoscopic remission were not significantly different between groups. In the maintenance trial, numerically, more patients in the 12 mg twice daily group were in clinical and endoscopic remission than the other groups. More AE occurred in the higher dose upadacitinib arms, but the majority were mild or moderate in severity. The most serious AE occurred in the 12 mg twice daily arm. The most frequent AE included worsening CD, urinary tract infection, nausea/vomiting, and headache. Further studies of upadacitinib in CD are in progress.46-50
IL-23 INHIBITOR
Guselkumab
Whereas ustekinumab blocks both IL-12 and IL-23 by inhibiting their shared p40 subunit, guselkumab is a more selective antagonist, targeting the p19 subunit of IL-23.51 IL-23 activates the JAK-signal transducer and activator of transcription pathways (Figure 1).51 The Th17 pathway is subsequently activated, leading to further production of cytokines such as IL-17, IL-21, IL-22, and IL-26. Guselkumab has already demonstrated efficacy without any serious AE and has been approved for moderate-to-severe plaque psoriasis. No clinical trials have been published yet on this agent; however, one published case report described a female with CD who achieved deep remission with guselkumab.52 Clinical trials are underway and planned for study in UC and CD.53-56
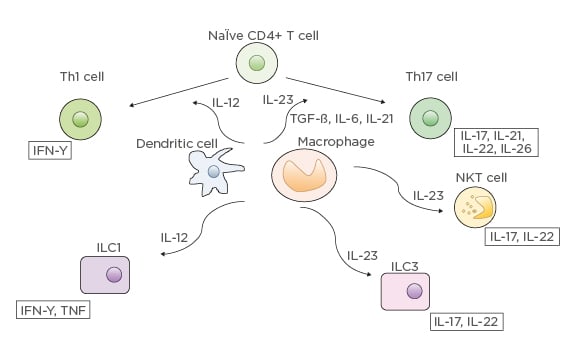
Figure 1: IL-12 and IL-23 differentiation pathways.
CD: cluster of differentiation; ILC: innate lymphoid cells; NKT: natural killer T.
From “Anti-interleukin-23 agents for the treatment of ulcerative colitis” by Jurij Hanžel & Geert R. D’Haens, Expert Opinion on Biological Therapy, published April, 2020, reprinted by permission of the publisher (Taylor & Francis Ltd).
hl][Mirikizumab[/hl]
Like guselkumab, mirikizumab is a monoclonal antibody that binds the p19 subunit of IL-23. Mirikizumab was studied in a Phase II trial in moderate-to-severe UC.17 Patients were randomised to one of three doses of intravenous mirikizumab or placebo. Those that responded to mirikizumab at Week 12 were randomised to 200 mg subcutaneous mirikizumab every 4 or 12 weeks through to Week 52. Those that responded to placebo at Week 12 were continued on placebo through to Week 52.
Compared to placebo, more patients in the mirikizumab arms were in clinical remission at Week 12, the primary endpoint; however, these differences were nonsignificant. Significantly more patients receiving mirikizumab had a clinical response by Week 12 compared to placebo. Significantly more patients in the 50 mg and 200 mg arms achieved endoscopic remission by Week 12. In the maintenance trial, by Week 52, 53.7% and 39.7% of patients given mirikizumab every 4 and 12 weeks, respectively, were in clinical remission, with similar rates between biologic-naïve and biologic-exposed patients. In the induction trial, the most frequent AE were nasopharyngitis, worsening of UC, and anaemia. In the maintenance trial, the most frequent AE were nasopharyngitis, upper respiratory tract infection, arthralgia, and influenza. Further trials are underway studying mirikizumab in UC57-59 and CD.60-62
Risankizumab
Risankizumab also targets the p19 subunit of IL-23. A Phase II, 12-week induction study in patients with moderate-to-severe CD compared intravenous risankizumab to placebo.18 Patients were randomised to either 200 mg or 600 mg of intravenous risankizumab or placebo, each administered every 4 weeks. Significantly more patients in the risankizumab groups achieved clinical remission at Week 12, the primary endpoint. Higher numbers of patients in the 600 mg compared with placebo group achieved clinical response, endoscopic remission, and deep remission. Mucosal healing, defined as absence of mucosal ulceration, was not achieved at higher rates in the risankizumab groups compared with placebo. Serious AE included worsening of CD and infections (n=3 [risankizumab groups]; n=3 [placebo group]).
Results from an open-label extension study were subsequently published.19 Of the 44 patients in the open-label extension arm, 71% were in clinical remission at Week 52 and 81% demonstrated a clinical response. Further studies on risankizumab are underway in UC63,64 and CD.65-67
Anti-α4β7 and Anti-αeβ7 Integrin Monoclonal Antibody
Etrolizumab
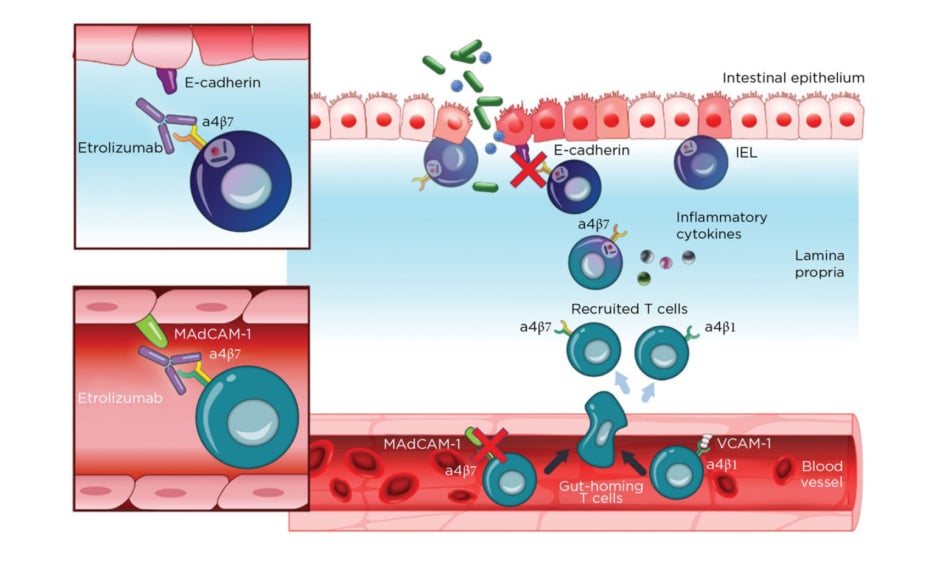
Etrolizumab is a monoclonal antibody against α4β7 and αEβ7 integrin and thereby prevents leukocyte binding with MAdCAM-1 and E-cadherin, respectively (Figure 2).37 After promising Phase I results,20the Phase II trial (EUCALYPTUS) results were published.68
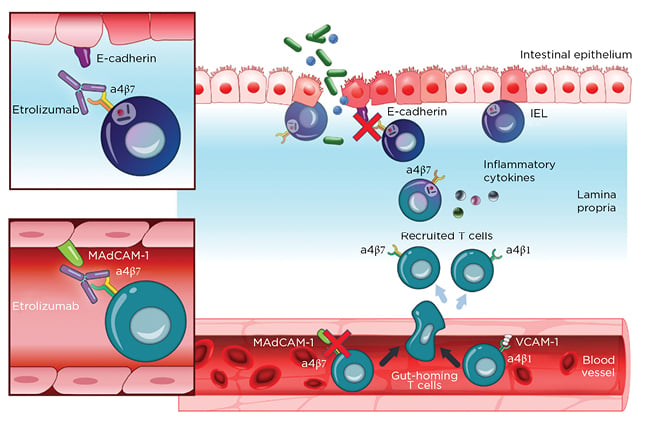
Figure 2: Etrolizumab mechanism of action.
IEL: Intraepithelial lymphocytes; MadCAM-1: mucosal addressin cell adhesion molecule 1; VCAM-1: Vascular cell adhesion protein 1.
From Sandborn et al.,37 2020. Creative Commons 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/).
In EUCALYPTUS, patients underwent randomisation with assignment to one of two doses of subcutaneous etrolizumab or placebo.
At 10 weeks, statistically more patients in the etrolizumab arms were in clinical remission compared to the placebo arm, the primary endpoint (21%, 10%, and 0% in the 100 mg etrolizumab, 300 mg etrolizumab plus loading dose etrolizumab, and placebo group, respectively, [comparisons p=0.0040 and p=0.048]). No significant differences were reported for clinical remission at Week 6, clinical response at Week 10, mucosal healing (defined as a Mayo Endoscopic Subscore 0 or 1) at Week 10, or histopathologic disease severity score. Numerically, more AE were reported in the placebo group. One serious infection was reported in the placebo group.
Further studies are underway, including open-label extension studies in UC (SPRUCE,69 HIBISCUS I,70HIBISCUS II,71 LAUREL,72 GARDENIA,73 HICKORY,74 COTTONWOOD75) and CD (BERGAMOT,76JUNIPER77).21
Sphingosine-1-Phosphate Subtypes 1 and 5 Modulator
Ozanimod
Ozanimod is an oral small molecule that modulates sphingosine-1-phosphate 1 and 5 receptor subtypes. Sphingosine-1-phosphate subtype 1 has been demonstrated to play a key role in lymphocyte trafficking; when antagonised, lymphocytes are no longer able to travel from secondary lymphoid organs into the circulation.78 The TOUCHSTONE Phase II trial studied ozanimod in moderate-to-severe UC.22 In the induction phase, patients were randomised to one of two doses of ozanimod or placebo for 8 weeks. Patients who responded in the induction trial continued, blinded, on the medication for 24 weeks. Those that did not respond during the induction trial were given the option to continue open-label treatment.
Significantly more patients in the 1 mg/day of ozanimod arm achieved clinical remission, the primary outcome, at Week 8 compared to placebo (16% versus 6%; p=0.048). Numerically, more patients in the 0.5 mg/day of ozanimod arm achieved clinical remission at Week 8 compared to placebo (14% versus 6%; p=0.14). Significantly more patients in both ozanimod arms demonstrated mucosal healing and Mayo Endoscopic Subscore 0 or 1 at Week 8, compared to placebo. Numerically, more patients in the ozanimod arms demonstrated histologic remission at Week 8 compared to placebo.
At Week 32, significantly more patients receiving 1 mg or 0.5 mg/day of ozanimod remained in clinical remission compared to placebo (21%, 26%, and 6%, respectively; p=0.01 and p=0.002 compared to placebo). Mucosal healing and histologic remission at Week 32 were achieved in significantly more patients in both ozanimod arms compared to placebo. More AE were reported in the placebo arm, including serious AE and AE leading to drug discontinuation.
Preliminary results from the Phase III trial studying ozanimod in UC79 have been reported as promising, with significantly increased numbers of patients achieving clinical remission after induction therapy at Week 10 and maintaining remission up to Week 52;80 published results are pending. Further studies are underway with ozanimod in UC81-84 and CD.85,86
Mesenchymal Stem Cells
Mesenchymal stem cells (MSC) are adult stem cells which lack the immunogenicity required for preconditioning regimens. Trials have been performed using intrafistular autologous adipose-derived stem cells (ASC),23 intrafistular bone marrow-derived mesenchymal stem cells (bmMSC),24 and intravenous bmMSC in both fistulising25 and luminal CD26 with mixed results. Numerous future studies are underway to evaluate the role of MSC in both CD and UC.87-90
CONCLUSION
Here, the authors have given an overview of novel and emerging therapies for use in the management of CD and UC. Given the rates of induction of remission and maintenance of remission with current therapies, future agents with new mechanisms of action are needed. It is promising that primary endpoints of clinical remission are reported in >50% of patients with agents such as mirikizumab and guselkumab, higher rates than achieved by agents currently approved for CD and UC. One network meta-analysis analysing rheumatoid arthritis data suggests that filgotinib is more effective than tofacitinib, with upadacitinib following tofacitinib.91,92
Future studies are also required to gain additional knowledge regarding positioning of agents. Personalised medicine or matching optimal medical therapies to patients based on their individual characteristics requires further study, although some predictive characteristics are emerging.93,94 To date, the only head-to-head trial of biologics compared vedolizumab and adalimumab (the VARSITY trial) and the authors of the study concluded that vedolizumab outperformed adalimumab.95 In addition to efficacy, the safety profiles of each medication will play a large role in the selection of agents for each patient. Targeted therapies with higher specificty are potentially safer than existing therapies; for example JAK-1 inhibitors have been compared in the rheumatology literature to tofacitinib, a nonselective JAK inhibitor.96 Furthermore, medications with gut selectivity are likely to have more acceptable long-term safety profiles. Therapies that can be topically administered, such as MSC, are likely to play a significant role in the future management of perianal fistulising disease in CD. Overall, these new therapies for the management of IBD are exciting and are likely to help more patients achieve induction and long-term maintenance of remission with fewer AE.