Meeting Summary
This article summarises an Orchard Therapeutic-sponsored symposium titled ‘New horizons for metachromatic leukodystrophy – with the advent of newborn screening’, which was delivered on 5th September 2024 as part of the Society for the Study of Inborn Error of Metabolism (SSIEM) annual congress in Porto, Portugal.
During the symposium, the panellists discussed the applicability of the Wilson and Jungner criteria to metachromatic leukodystrophy (MLD), which they considered a strong candidate for newborn screening (NBS) thanks to the existing supporting evidence. This includes the availability of a screening test, the agreement on how to confirm diagnosis after positive screening, the presence of a prospective population-based newborn screening project that identified at least one infant with the condition, and the evidence that an early identification through NBS leads to better health outcomes.
In the symposium, the speakers also reminded the audience of the existence of a validated three-tier screening algorithm of recent publication and the availability of two consensus guidelines that have been published in both the EU and the USA, and which unanimously support the implementation of NBS for MLD.
Introduction
The decision of whether or not to screen for a disease still relies heavily on a framework developed almost 40 years ago, when the WHO commissioned a report on screening from James Maxwell Glover Wilson, then Principal Medical Officer at the Ministry of Health in London, UK, and Gunner Jungner, then Chief of the Clinical Chemistry Department of Sahlgren’s Hospital in Gothenburg, Sweden. The report, published in 1968, was titled ‘Principles and practice of screening for disease and it has since become a public health classic’.1
Despite the admirable method of combating disease, the practice of screening comes with some challenges, and the authors were preoccupied with the notion that: “The central idea of early disease detection and treatment is essentially simple. However, the path to its successful achievement (on the one hand, bringing to treatment those with previously undetected disease, and, on the other, avoiding harm to those persons not in need of treatment) is far from simple, though sometimes it may appear deceptively easy.”1
The Wilson and Jungner Criteria and Newborn Screening
For this reason, Wilson and Jungner attempted to define screening criteria to guide the selection of conditions that would be suitable for screening, based, among other factors, on the capacity to detect the condition at an early stage and the availability of an acceptable treatment:
1. The condition sought should be an important health problem.
2. There should be an accepted treatment for patients with recognised disease.
3. Facilities for diagnosis and treatment should be available.
4. There should be a recognisable latent or early symptomatic stage.
5. There should be a suitable test or examination.
6. The test should be acceptable to the population.
7. The natural history of the condition, including development from latent to declared disease, should be adequately understood.
8. There should be an agreed policy on whom to treat as patients.
9. The cost of case-finding (including diagnosis and treatment of patients diagnosed) should be economically balanced in relation to possible expenditure on medical care as a whole.
10. Case-finding should be a continuing process and not a ‘once and for all’ project.
These criteria are particularly challenging when applied to rare diseases, conditions affecting less than 1 in 2,000 people.2 It is currently estimated that there are over 7,000 rare diseases, and while 80% of rare diseases have an identified genetic origin, they can also be caused by disordered immunity, infections, allergies, deterioration of body tissues and organs, or disruption to development while in the womb.
MLD is a rare inherited lysosomal storage disease affecting 1 in 100,000 newborns and caused by deficiency of arylsulfatase A (ARSA), due to mutations in the ARSA gene.3 Reduced ARSA activity results in the accumulation of sulfatides in the CNS and peripheral nervous system, leading to progressive demyelination, neuroinflammation, and neurodegeneration.4-6 These events result in progressive motor and cognitive deterioration, with loss of motor and neurocognitive functions, and ultimately death.4,5,7 Three clinical forms are commonly described on the basis of age at first symptom onset: late-infantile (LI; ≤30 months), juvenile (subdivided into early juvenile [30 months–6 years] and late juvenile [7–16 years]), and adult MLD (≥17 years), with earlier age at onset or presence of motor symptoms at onset associated with a more severe and rapid disease course.4-6,8,9 Regardless of the clinical variant, the underlying disease pathophysiology is similar for all phenotypic forms of MLD.4,7,10
The fast progression of the LI subtype of the disease (accounting for the majority of the MLD cases, or about 60% of the affected population), clearly highlights the importance of identifying children with urgency. Affected children initially show a normal development, but once symptoms start to become evident, patients may no longer be eligible for treatment, entering a disease progression phase that leads to irreversible and progressive neurological damage and ultimately death, usually in the first decade of life.11
The early detection of the disease during the asymptomatic phase is therefore paramount to increase the chances of a timely intervention and better clinical outcomes. Should MLD be considered a good candidate for newborn screening, then? As explained by Amy Gaviglio, a Genetic Counsellor, Public Health Genetics and Rare Disease Consultant from Minneapolis, Minnesota, USA, with a wealth of experience in the expansion of newborn screening programmes, to answer this question, we must first consider several criteria: 1) Is there a screening test available for use at a population level in the newborn period? 2) Is there an agreed-upon way for a clinical specialist to confirm the diagnosis after a positive screening? 3) Is there a prospective, population-based newborn screening project that has identified at least one infant with the condition? 4) Does early identification through newborn screening lead to better health outcomes compared to usual clinical identification?
The Principles and Practice of Newborn Screening
As for any disease, the inclusion of MLD on the newborn screening panel requires the knowledge and the generation of evidence to confirm its readiness for population newborn screening. To achieve this goal, an international group of scientists, patient advocates, and MLD experts started three key retrospective studies, using dried blood spots to validate the screening algorithm.12-14 The promising results, data collected, and knowledge acquired led to the initiation of 11 additional prospective investigator-initiated newborn screening studies for MLD, which are active throughout the USA, Europe, and the Middle East, with more than 300,000 newborns screened to date (as of September 2024). Most importantly, through these prospective studies, five newborns with no known previous MLD family history were screened positive and subsequently diagnosed with the disease. The evidence generated is being used to support the nomination to add MLD to the newborn screening panel in Europe (including the UK, Ireland, France, and Germany) and in the USA, and Norway is the first country in the world to add MLD to the national newborn screening programme, officially starting on 6th January 2025.
Lucia Laugwitz, a clinician scientist and child neurologist in training in the Department of Neuropediatrics and the Institute for Medical Genetics and Applied Genomics at the University of Tuebingen, Germany, explains that when starting a newborn screening, the first and most important aspect is to establish the existence of a screening test available for use at a population level in the newborn period. In 2024, Laugwitz et al.15 validated and published the results of a three-tier screening algorithm for MLD; the protocol includes two biochemical tests (sulfatides and enzyme activity) followed by a third genetic test not only for the ARSA gene, but also the SUMF1 and PSAP genes associated with multiple sulfatides deficiency and prosaposin B deficiency respectively.
Due to the subtleness of MLD, it is important that all three genes are tested, because, at a biochemical level, both SUMF1 and PSAP can mimic MLD but have currently no approved treatments. The combination of two sulfatides (C16:0 and C16:1-OH) is used to reduce the number of false positives in the first tier, and the subsequent need for second-tier enzymatic biochemical testing, which in turn improves the feasibility for implementation at a national level (Figure 1).
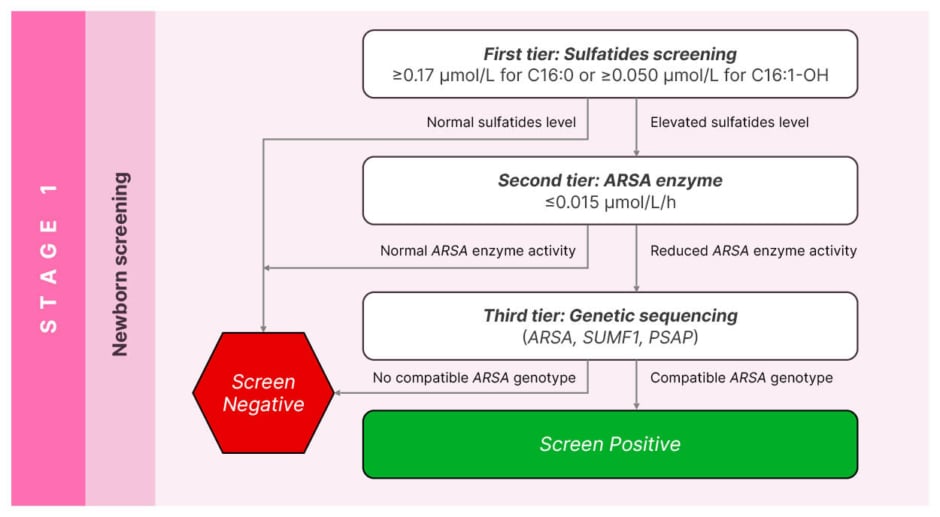
Figure 1: Validated screening algorithm for metachromatic leukodystrophy.15
Adapted from Laugwitz et al.15
Selecting Conditions Suitable for Newborn Screening
In 2024, two complementary consensus guidelines on MLD have been published: one from the MLD initiative (MLDi)16,17 stressing the importance, among other things, of implementing newborn screening to promptly identify children and ensure proper management and care; and one in the USA with a prominent focus on patient care and specific monitoring requirements.18 As highlighted by Laura Adang, Assistant Professor of Neurology at the Children’s Hospital of Philadelphia, Pennsylvania, USA, the most relevant aspect of consistency between the two documents is that all the authoring experts unanimously support the implementation of newborn screening for MLD, whose subacute nature makes it difficult to be detected until children become symptomatic, affecting their ability to potentially receive appropriate treatment. The same experts also agree and strongly recommend initiating treatment in identified individuals before symptom onset, the best option to help the patients and their families.
As clearly shown in Figure 1, the screening algorithm includes a biochemical confirmation of ARSA enzyme activity in leukocytes and urinary sulfatides (performed in MLD expert centres) followed by genetic confirmation, which is required to help predict MLD subtypes (family history, genotype, and ARSA enzyme activity). In general, 80% of the time experts anticipate what MLD subtype the child will have; therefore, the consensus guidelines are particularly relevant to support the management and monitoring of the 20% of the patients with uncertain subtypes, with the goal of early intervention.
There are several known and recognised challenges, in particular, genetic variants of unknown significance and pseudo deficiencies; their existence is not dismissed by the expert’s community, who recognise the need to establish essential and wider collaboration. With time, the guidelines will be refined and updated to reflect new discoveries; gradually, new variants of unknown significance will emerge once national newborn screening programmes are implemented, and in parallel, the understanding of the entire MLD community of clinicians, scientists, and experts will improve, leading to better patient care and treatment outcome, so long as the information is shared as openly as possible.
Laugwitz et al.17 were not only able to implement a newborn screening algorithm but also a comprehensive care pathway going from the confirmation of MLD diagnosis to the clinical assessment and subtype prediction and treatment decision. Screening is only the first step of this process, but how should the identified MLD children be managed? Is early identification through newborn screening the necessary step leading to better health outcomes compared to clinical identification?
In a paper published by Claire Horgan, Senior Clinical Research Fellow in paediatric bone marrow transplant, CAR-T, and stem cell gene therapy at Royal Manchester Children’s Hospital in Manchester, UK, it was reported that in the year following NHS approval of an active treatment for MLD, 17 UK patients with MLD were referred for treatment.19 Four patients met eligibility criteria and were treated, whereas 11 patients failed screening; 10 due to symptomatic disease (LI subtype) and one with early juvenile disease and cognitive decline. Two further patients with later onset subtypes did not meet the approval criteria, and three out of four treated patients were diagnosed by screening after MLD was diagnosed in a symptomatic older sibling. This result is a clear testament to the challenges of diagnosing MLD in a timely manner before symptom onset, which further supports the need for newborn screening.
The key to progress in this field is cooperation within the expert’s community. In Europe, the establishment of a network of MLD experts under the umbrella of the MLDi is an example of this cooperation. The MLDi is an international patient registry for MLD and an academic collaborative network, and data in the MLDi registry can be used for academic research, regulatory decision-making, and drug development. In the USA, the establishment of the Global Leukodystrophy Initiative Clinical Trials Network (GLIA-CTN), a consortium of scientists, industry stakeholders, and patient advocacy leaders, promotes advances in the diagnosis and treatment of leukodystrophies, and specifically, it seeks to create a robust research infrastructure that will allow for collection and analysis of longitudinal natural history data, development of novel clinical outcome assessments, and identification of surrogate biomarkers, ultimately paving the way for transformative therapeutic trials across the leukodystrophies.
Conclusion
MLD strongly fulfils the Wilson and Jungner criteria for newborn screening due to the availability of a screening test for use at a population level in the newborn period, the availability of an agreed way for a clinical specialist to confirm the diagnosis after a positive screen, the presence of a prospective, population-based newborn screening project that has identified at least one infant with the condition, and the potential of better health outcomes of an early identification through newborn screening compared to usual clinical identification.
When thinking about diseases and what makes them good candidates for newborn screening, it is paramount they meet certain criteria, including the feasibility of testing for that disease in the newborn period. Typically, this is done for the purpose of newborn screening using a dried blood spot matrix on dried blood that is collected from the newborn around 24–72 hours after birth, while ensuring that that test has low false positive rates and low false negative rates, as well. A second important criteria to consider is whether the natural history of the disease and the type targeted with newborn screening are sufficiently understood. MLD is a spectrum; therefore, it is fundamental to recognise what that spectrum looks like and how to target the various subtypes that may have effective treatments. The third condition considers the feasibility of the test, to make sure the process works. For this reason, prospective pilot studies that look at the feasibility of this testing all the way through the diagnosis and administration of early pre-symptomatic interventions are exceptionally useful. Lastly, the existence of an effective treatment, which is needed asymptomatically to improve outcomes, completes the picture.
For a family, hearing that their child has a fatal disorder is extremely difficult, and they require appropriate social and psychological support, explained Hanka Dekker, Director of VKS, Zwolle, the Netherlands. In certain situations, and until now, parents had to care for both an index patient and their younger sibling, both diagnosed with the disease, witnessing their children losing all their abilities within years. In many cases, these challenges led to the family break-up. Newborn screening can change all this by identifying the index patient early enough to be treated, and the resulting psychological follow-up for these families will be much better.
As explained by Gaviglio, when considering these criteria and applying them to MLD, it is appropriate to say that the disease meets them all and should be included in newborn screening panels. This will help the early identification of patients with MLD while still in the asymptomatic phase, allowing for early access to potential disease-modifying treatments and improved health outcomes that would otherwise be negated, leading to irreversible neurodegeneration and early death of affected children.
IE-NoP-2500009
March 2025






