Abstract
Renal cell carcinoma (RCC) is the most common primary tumour of the kidney. RCC is a clinically and pathologically heterogenous entity, which has traditionally been classified under two broad categories: clear-cell and non-clear cell. With improved molecular diagnostic methodologies and genetic testing, the classification of RCC has shifted from a morphological basis to a molecular/genetic focus, and has been systematically updated to reflect these advancements. The new 2022 World Health Organization (WHO) classification of RCC is the most recent of these updates, and contains significant changes, as compared to the previous 2016 classification. The most substantial of these changes is the establishment of a new category of molecularly-defined RCC, including TFE3-rearranged RCC, TFEB-altered RCC, ELOC-mutated RCC, fumarate hydratase-deficient RCC, succinate dehydrogenase-deficient RCC, ALK-rearranged RCC, and SMARCB1-deficient renal medullary carcinoma. In this narrative review, the authors briefly summarise the histopathological characteristics, clinical course, current treatment standards, and future treatment directions of each of these molecularly-defined RCC subtypes.
Key Points
1. This article summarises the key pathologic and clinical characteristics of the molecularly-defined renal cell carcinoma (RCC) subtypes incorporated within the new 2022 World Health Organization (WHO) classification system. This change signifies a broader trend in oncology, emphasising the role of molecular and genomic tumour descriptions.2. Molecularly-defined RCC subtypes may pose diagnostic challenges, due to morphological variability. Ancillary techniques, including fluorescence in situ hybridisation, immunohistochemistry, and next-generation sequencing, are essential for accurate identification, requiring pathologists to adapt to this molecular-focused approach.
3. The clinical impact of molecularly-defined RCC subtypes remains uncertain. Interdisciplinary collaboration is emphasised for integrated diagnosis and treatment. Future research should focus on elucidating molecular pathways, exploring targeted therapies, and conducting prospective trials for personalised treatment approaches.
INTRODUCTION
Renal cell carcinoma (RCC) is the most common primary tumour originating from the kidney, and accounts for more than 90% of all renal malignancies.1 RCC is a clinically and pathologically heterogenous entity, which can arise from various cell types within the nephron. RCC has been traditionally classified into two broad pathologic categories: clear cell RCC (ccRCC) and non-clear cell RCC. Under the non-clear cell umbrella, the two most common histologic subtypes are papillary RCC (PRCC) and chromophobe cell RCC.
Historically, RCC classification has been primarily morphologically focused, but has more recently incorporated a molecular/genetic basis.2 In 2016, the World Health Organization (WHO) classified RCC according to the latest knowledge about genetics, pathology, and epidemiology.3 Subtypes were named largely on the basis of cytologic features (e.g., chromophobe RCC), cell of origin within the nephron (e.g., collecting duct RCC), architectural features (e.g., papillary RCC), pathognomonic molecular/genetic alterations and mutations (e.g., succinate dehydrogenase [SDH]-deficient RCC), and association with specific renal diseases (e.g., acquired cystic disease-associated RCC).3
With the advancement and widespread adoption of genomic sequencing, diagnostic methodology has shifted further from morphology to molecular descriptions.4 In 2022, the WHO introduced a new classification system consistent with this paradigm shift.4 This new system focuses more on a molecularly-driven tumour classification, and introduces several new molecularly-defined renal tumours (Table 1). The 2022 classification represents the first comprehensive molecular classification of renal tumours.4,5 The new subtypes include: TFE3-rearranged RCC, TFEB-altered RCC, ELOC-mutated RCC, fumarate hydratase (FH)-deficient RCC, SDH-deficient RCC, ALK-rearranged RCC, and SMARCB1-deficient renal medullary carcinoma (RMC).6
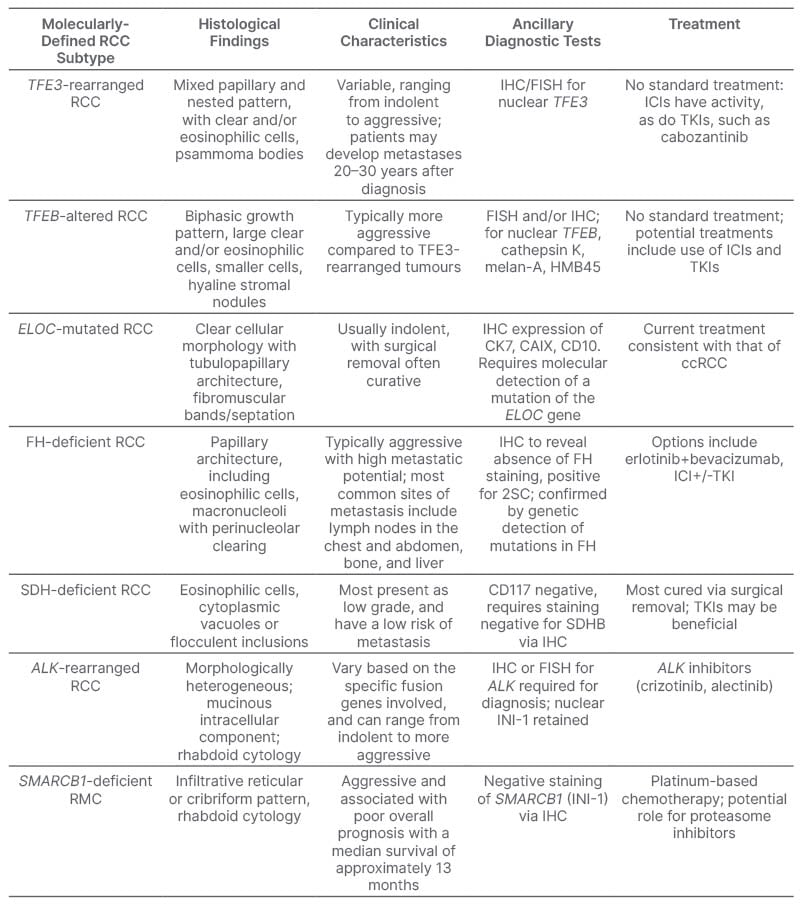
Table 1: Summary of molecularly-defined renal cell carcinoma subtypes.
ccRCC: clear cell renal cell carcinoma; FH: fumarate hydratase; FISH: fluorescence in situ hybridisation; IHC: immunohistochemistry; RCC: renal cell carcinoma; RMC: renal medullary carcinoma; SCH: succinate dehydrogenase; TKI: tyrosine kinase inhibitor; 2SC: 2-succinocysteine.
Molecularly-defined RCC may demonstrate heterogeneous morphology or architecture, and cannot necessarily be diagnosed by morphology alone.4 Accurate pathologic diagnosis requires a high degree of suspicion based on subtle morphologic features. Hence, these tumours require the use of ancillary techniques, such as fluorescence in situ hybridisation (FISH), immunohistochemistry (IHC), and next-generation sequencing, for their identification and diagnosis.6
It is important to note that some of the aforementioned molecularly-defined RCC, such as FH-deficient RCC, are derived from the previous classification of Type 2 PRCC, which has since been removed in the 2022 classification.7 Due to their morphological variability, these subtypes have been segregated from this previous morphologic label, and what was known formerly as Type 1 PRCC is now referred to as classical PRCC.7
From a clinical perspective, it is yet to be determined how these molecularly-defined renal tumours will impact clinical management. The molecular classification of other tumour types, such as lung cancer, breast cancer, and melanoma, has made a significant impact on the treatment and therapeutic landscape of these tumours, including the development of targeted therapy specific to certain molecular alterations.7-10
This narrative review aims to describe the characteristics of each of the new molecularly-defined subtypes of RCC, as well as to discuss the potential therapeutic implications of this reclassification.
TFE3-REARRANGED RENAL CELL CARCINOMAS
TFE3-rearranged RCC is most commonly discovered during childhood, and is found in up to 75% of childhood RCC.6 A history of previous chemotherapy is present in up to 15% of cases.9 In previous classifications, TFE3-rearranged RCC was organised under microphthalmia transcription (MiT) family translocation carcinomas, along with TFEB-rearranged RCC.10 As its name indicates, TFE3-rearranged RCC is characterised by a translocation of TFE3, which is derived from the MiT subfamily of transcription factor genes.10 The Xp11 translocation is pathognomonic for TFE3-rearranged RCC.6 Further, TFE3 is notably able to rearrange with more than 20 gene partners to create fusion subtypes, with variable tumour morphology and clinical behaviour.11
Histologically, most cases reveal a mixed papillary, solid, and nested pattern, with large voluminous clear and eosinophilic cells, and psammoma body calcifications may be present.10,12 IHC expression of melanocytic markers (HMB45, Melan-A) is present in up to 40% of cases.10 Due to its heterogeneity, and resemblance to other types of RCC, however, IHC for nuclear expression of TFE3 and FISH assay for TFE3 rearrangement are required for accurate diagnosis.6,12
The clinical behaviour of TFE3-rearranged RCC can be variable, ranging from indolent to highly aggressive.6 Additionally, some patients may develop metastases 20–30 years after diagnosis.12 There are no standard treatment recommendations for advanced/metastatic TFE3-rearranged RCC, and most treatment regimens are consistent with those used in ccRCC.13 A retrospective study has suggested that immune checkpoint inhibition is effective in the treatment of advanced TFE3 RCC, as well as specific tyrosine kinase inhibitors (TKI), such as cabozantinib.6,12
TFEB-ALTERED RENAL CELL CARCINOMA
TFEB-altered RCC is similarly predominantly found in children, but is less common than TFE3-rearranged RCC.6 There are two primary types of TFEB-altered RCC: TFEB-rearranged RCC and TFEB-amplified RCC.12 The TFEB gene is located on chromosome six and, in TFEB-rearranged RCC, is most commonly translocated to chromosome 11, where it fuses with the MALAT1 gene.10 As mentioned previously, TFEB-rearranged RCC was also previously organised under the MiT family translocation carcinomas.10 In TFEB-amplified RCC, the elevated expression of the TFEB gene often coexists with the amplification of other oncogenic genes, such as vascular endothelial growth factor A.12
The histologic appearance of TFEB-altered RCC is variable, with solid and papillary architecture.12 The most important morphologic clues are a biphasic growth pattern of large, clear, and eosinophilic cells with high nuclear grade, and compact collections of smaller tumour cells clustered around nodules of hyaline basement membrane-type stromal material.12 Melanocytic markers and cathepsin K are positive in up to 90% of cases.10 Definitive diagnosis of TFEB-altered RCC requires the use of IHC for nuclear TFEB expression, and FISH assay for TFEB-rearrangement versus amplification.12
Clinically, TFEB-amplified RCC is thought to be more aggressive compared to TFE3-rearranged tumours, with a 5-year survival rate of 48%.12 Given its rarity, there are a lack of current treatment recommendations for TFEB-altered RCC.6,12 TFEB-altered RCC can be misdiagnosed as conventional ccRCC; hence, prescribed treatment is often that of ccRCC.12 Some studies indicate that TFEB may evade the immune system via programmed cell death ligand 1 expression; hence, immune checkpoint inhibitors (ICI) may be a therapeutic option.6 Similarly, targeted therapies against vascular endothelial growth factor receptors may be effective in the treatment of TFEB-amplified RCC.6
ELOC (FORMALLY TCEB1)- MUTATED RENAL CELL CARCINOMA
ELOC-mutated RCC is an uncommon malignancy that predominantly affects older age groups, as compared to the translocation RCC subtypes.12 The ELOC gene codes for the protein elongin C, an important component of the von Hippel–Lindau protein complex, and is responsible for the ubiquitination and degradation of HIF1α.14 With an ELOC mutation, this degradation is inhibited, leading to HIF1α accumulation, and activation of various oncogenes with subsequent tumourigenesis.14
Morphologically, ELOC-mutated RCC resembles ccRCC with predominant voluminous clear cell cytology, acinar or tubulopapillary architecture, and smooth muscle-rich fibromuscular bands/septation.12 The tumour is, in most cases, diffusely positive for CD10, CK7, and CAIX, and needs therefore to be distinguished from the clinically indolent clear cell papillary renal cell tumour.12 The staining for CAIX is a box-like pattern, rather than the cup-shaped pattern seen in clear cell papillary renal cell tumour.15
This entity has, in the past, been described as ‘RCC with fibromuscular stroma’.7 It is important to note, however, that this morphological presentation is not pathognomonic, as there exists some RCC with fibromuscular stroma caused by mTOR pathway mutations, and these are histologically indistinguishable from ELOC-mutated RCC.7 Therefore, ELOC-mutated RCC requires molecular characterisation of a mutation of the ELOC gene for definitive diagnosis.6,13,14
The current treatment of ELOC-mutated RCC is consistent with that of ccRCC, as ELOC-mutated RCC was previously considered to be one type of ccRCC.12 Clinically, ELOC-mutated RCC tends to be more indolent, and surgical removal in localised disease is often curative.6
FUMARATE HYDRATASE-DEFICIENT RENAL CELL CARCINOMA
FH-deficient RCC is a rare form of RCC that predominantly affects adults; it was previously classified as hereditary leiomyomatosis and RCC syndrome-associated RCC.6,12 FH is an enzyme in the tricarboxylic acid cycle that is involved in the production of cellular energy through oxidative phosphorylation.12,16 Autosomal dominant germline mutations in the gene encoding FH can cause accumulation of fumarate, which has oncogenic properties, and can drive tumourigenesis.16 The primary rationale for the reclassification of hereditary leiomyomatosis and RCC-syndrome associated RCC is the discovery of somatic biallelic alterations causing sporadic cases of FH-deficient RCC, in addition to the more common hereditary form.6 Positive family history, and multiple cutaneous and (in females) uterine leiomyomas are not always present.17 Genetic testing of family members for FH mutation is necessary, due to up to a 30% lifetime risk of RCC in affected individuals.17
Prior to the discovery of FH-deficient RCC, these tumours were generally classified as high grade RCC (not otherwise specified), Type 2 papillary RCC, or collecting duct carcinoma. FH-deficient RCC may display a variety of histologic growth patterns, the most common being papillary, with other patterns being solid, tubulocystic, and cribriform or sieve-like.12 Tumour cells generally have granular eosinophilic, rather than clear, cytoplasm.12 A characteristic cytologic feature that is an important clue to raise suspicion of the diagnosis, and pursue appropriate confirmatory IHC testing, is the presence, at least focally, of very large ‘cherry red’ inclusion-like macronucleoli, sometimes with perinuclear clearing.12 IHC shows loss of cytoplasmic reactivity for FH, and strong nuclear and cytoplasmic staining for 2-succino-cysteine, due to the enzyme blockade in the tricarboxylic acid cycle.6,12
Clinically, FH-deficient RCC is often aggressive, with high metastatic potential.6 The most common sites of metastasis include lymph nodes in the chest and abdomen, bone, and liver.12 There is no clear standard of therapy for FH-deficient RCC, and current treatment is largely consistent with that of ccRCC.12 Several drugs have been explored in recent years, including ICIs (e.g., nivolumab and ipilimumab), and TKIs (e.g., sunitinib and pazopanib). Some data suggest that treatment with ICIs in combination with TKIs may be superior to monotherapy.18 Furthermore, a recent trial evaluating the combination of erlotinib and bevacizumab has shown favourable results, with a response rate of 51%.19
SUCCINATE DEHYDROGENASE-DEFICIENT RENAL CELL CARCINOMA
SDH-deficient RCC is predominantly found in male patients, and can affect individuals of all ages.12 SDH is a mitochondrial enzyme complex composed of four subunits (SDHA, SDHB, SDHC, and SDHD), which plays an important role in energy metabolism.20 The pathogenesis of SDH-deficient RCC involves germline mutations of the genes that encode these SDH subunits, most commonly in SDHB.12,20 These germline mutations result in the development of adrenal phaeochromocytomas, extra-adrenal paragangliomas, gastrointestinal stromal tumours, and pituitary adenomas, in addition to RCC.20
SDH-deficient RCC can be solid or cystic, and up to 30% can be multifocal and bilateral.20 Histologically, tumour cells have a uniform granular eosinophilic cytoplasm, and if the nuclear grade is low, a serious misdiagnosis of benign renal oncocytoma is possible.12,20 The growth pattern is generally sheet-like or papillary.20 High grade nuclei, necrosis, and sarcomatoid transformation are described.20 The most characteristic pathologic feature of SDH-deficient RCC is the presence of cytoplasmic vacuoles or flocculent inclusions, representing enlarged mitochondria.6,12 IHC is generally negative in tumour cells for CD117 (in contrast to oncocytoma); intratumoural CD117-positive mast cells may be prominent.20 Diagnosis of SDH-deficient RCC requires an absence of cytoplasmic staining in tumour cells for SDHB, using IHC.12
Clinically, most cases of SDH-deficient RCC present as low-grade, and have a low risk of metastasis.12 In these cases, most cases of localised disease can be cured via surgical resection.6 Those tumours that present as high grade have a higher risk of metastasis (up to 70%), and, in these cases, studies have shown that using TKIs may be beneficial.6,12
ALK-REARRANGED RENAL CELL CARCINOMA
ALK-rearranged RCC is a rare tumour, with incidence estimated to be between 0.1–0.6%.12 The ALK gene functions to regulate cell proliferation and promote cell motility; hence, when gene rearrangement occurs, it can lead to tumourigenesis.12 Similar to TFE3-rearranged RCC, ALK-rearranged RCC may form fusion genes with various partner genes.21 Underlying sickle cell trait is seen with the VCL-ALK gene fusion.22
ALK-rearranged RCC can be very morphologically heterogeneous, with the majority expressing various growth patterns, including tubulopapillary, cribriform, solid, and sarcomatoid.4 There can even be a metanephric adenoma-like morphology.21 High-grade pleomorphic nuclei are typically present, as well as large cells with eosinophilic cytoplasmic inclusions (rhabdoid morphology).21 Cytoplasmic vacuolation with a signet ring cell mucinous intracellular component and myxoid stroma are also typical features, giving morphologic overlap with mucinous tubular and spindle cell carcinoma.21 Prominent tumour-infiltrating inflammatory cells are seen with the TPM3-ALK gene fusion. IHC shows tumour cells are generally positive for PAX8, CK7, and AMACR (racemase), and there is retained nuclear INI1 in contrast to SMARCB1-deficient RCC.12,21,23 Positive ALK staining by IHC, or FISH for ALK rearrangement, is required for definitive diagnosis.21,23
Clinically, the behaviour of ALK-rearranged RCC can vary, based on the specific fusion genes involved, and can range from indolent to more aggressive.12 Given the rarity of this subtype, there is no standard therapy for advanced disease.6,12 Extrapolating from the treatment of ALK-rearranged lung cancer, there is interest in the use of targeted inhibition of ALK, and there are case reports showing effectiveness of ALK inhibition in ALK-rearranged RCC.22,24
SMARCB1-DEFICIENT RENAL MEDULLARY CARCINOMA
SMARCB1-deficient RMC, previously classified simply as RMC, is an aggressive cancer that is found almost exclusively in adolescents and young adults with sickle cell trait, sickle cell disease, or other haemoglobinopathies.25 Increased sickling of red blood cells causes regional ischaemia in the renal medulla, which is thought to be key to its pathogenesis.25 The disease is characterised by the loss of tumour suppressor gene SMARCB1 on chromosome 22.12
Morphologically, SMARCB1-deficient RMC can be quite heterogeneous, but most commonly exhibits a highly infiltrative reticular, microcystic, or cribriform pattern, with spaces of variable sizes.6 Rhabdoid cytology with prominent eosinophilic cytoplasmic inclusions, and high-grade nuclei are typical histologic features.12,26 In addition to this morphology, and its characteristic clinical course, the absence of nuclear staining for SMARCB1 (INI1 protein) via IHC is utilised for diagnosis.12 It should be noted that there is pathologic and clinical overlap with collecting duct carcinoma, as both entities arise within the renal medulla.27 In contrast to SMARCB1-deficient RMC, collecting duct carcinomas typically retain SMARCB1 expression.27
Clinically, SMARCB1-deficient RMC is highly aggressive, and is associated with a poor overall prognosis, with a median survival of approximately 13 months.6 The current standard therapy for advanced SMARCB1-deficient RMC is systemic chemotherapy, including platinum-based regimens.6 Recent studies have explored targeted therapies, such as proteasome inhibitors.6,12 A clinical trial of the proteasome inhibitor ixazomib, in combination with gemcitabine and doxorubicin, in patients with RMC, is currently enrolling patients.28
DISCUSSION
The revised pathologic classification of RCC represents a significant shift in oncology, prioritising molecular characteristics over traditional tumour morphology, and is a departure from the more simplified clear cell versus non-clear cell RCC classification. This is reflective of a broader trend in oncology, as molecularly-targeted therapies are increasingly showing promise. Current treatments for these newly-defined RCC subtypes are still, for the most part, extrapolated from treatments for ccRCC. Still, the hope is that a deeper understanding of these tumours at the molecular level will lead to more targeted and effective therapies. This classification is an important step forward in understanding the genetic and molecular basis of rare kidney cancer subtypes, as it recognises the biologic heterogeneity of RCC, and provides a more precise characterisation of these tumours.
This molecular classification emphasises the importance of ancillary diagnostic techniques, such as specialized IHC, FISH, and next-generation sequencing for diagnosis, as these molecularly-defined RCC subtypes may not exhibit distinct morphological features. Surgical pathologists need to have a high index of suspicion for ordering these additional diagnostic tests, in order to catch the rare positive cases that would be expected in routine practice. Any tumour showing morphology that would have in the past been classified as Type 2 papillary RCC, collecting duct carcinoma, or RCC not otherwise specified, should be considered for additional ancillary molecular and IHC testing. If rhabdoid tumour cell cytology is encountered, ALK and INI1 IHC stains are mandatory to investigate possibility of ALK-rearranged RCC and SMARCB1-deficient RMC, respectively.
The implications of this reclassification for clinical practice remain uncertain. Beyond improved diagnostic precision, it could pave the way for the development of biomarker-selected targeted therapies, akin to those seen with other cancers, such as breast, lung, and melanoma. In addition, accurate pathologic categorisation can have important implications on prognosis (e.g., SMARCB1-deficient RMC, which is associated with a particularly aggressive phenotype, and poor prognosis). It is also likely that interdisciplinary collaboration between pathologists, geneticists, and oncologists will become more critical, as diagnosis and treatment become more intertwined with molecular findings.
Future research should focus on further elucidating the molecular pathways of these RCC subtypes, and determining whether specific targeted therapies can be selected based on molecular classification. Although challenging due to the rarity of these conditions, prospective clinical trials designed to enrol patients based on their molecular subtype would help to generate more robust data on the benefit of novel therapeutics. Clinical trials in other rare subtypes of RCC have been performed, including RMC and papillary RCC, showing that it is possible to conduct trials in less common diseases.29
As we move forward in RCC classification, it is becoming more evident that the simple distinction between ccRCC and nccRCC may not adequately capture the true biologic and pathologic complexity of this condition, and that future treatments may become more tailored to the underlying tumour biology.






