Abstract
An acute necrotising myopathy is a distinct form of uncommon muscle disease characterised by the rapid advancement of weakness affecting the limbs, neck, pharyngeal, respiratory, and occasionally cardiac muscles. It frequently arises as part of idiopathic inflammatory myopathies, which include conditions like polymyositis, dermatomyositis, and inclusion body myositis. Anti-hydroxy-3-methylglutaryl-coenzyme A reductase represents an infrequent immune-mediated necrotising myopathy. This case study involves a 55-year-old African American woman experiencing muscle weakness and recurrent falls over 2 months with elevated creatine kinase levels, indicating an inflammatory disease process. The patient received symptomatic management after excluding any critical illness. After initial treatment, she underwent outpatient follow-up along with necessary investigations that led to the definitive diagnosis.
Key Points
1. Statins, the most commonly prescribed medication, are linked to anti-hydroxy-3-methylglutaryl-coenzyme A reductase-induced myopathy. Early recognition is crucial due to its significant impact on muscle function.
2. This case report describes the diagnostic challenges and immunotherapy in a 55-year-old woman with anti-hydroxy-3-methylglutaryl-coenzyme A reductase immune-mediated necrotising myopathy.
3. Early statin discontinuation and combination immunotherapy improve outcomes in necrotising autoimmune myopathy, with multi-agent therapy being more effective in resistant or complex cases.
INTRODUCTION
Inflammatory myopathies constitute a wide range of diseases characterised by muscle inflammation and weakness. Dermatomyositis and polymyositis are the most widely recognised and extensively researched among these conditions.1 However, recent progress in medical research has revealed new distinct inflammatory myopathies,2 emphasising the complexity and diversity of these disorders. One such ailment is immune-mediated necrotising myopathy, also known as necrotising autoimmune myopathy (NAM), a form of autoimmune myopathy characterised by the acute or sub-acute onset of severe, symmetrical weakness primarily affecting the proximal musculature. It includes a rare subset associated with antibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR).3
Idiopathic inflammatory myopathies affect individuals across different ages and ethnicities, although they are generally uncommon. These myopathies feature persistent muscle inflammation, leading to progressive muscle weakness and disability. Identifying myositis-specific autoantibodies has improved diagnostic accuracy, enabling better categorisation and comprehension of these illnesses. Despite their overall rarity, their impact on patients’ lives is profound, demanding precise diagnosis and effective treatment strategies.
Anti-HMGCR myopathy poses unique challenges in clinical practice due to its infrequency as well as its potential association with statin use, commonly prescribed for hyperlipidaemia and cardiovascular risk reduction, the aspect known as statin-induced necrotising autoimmune myopathy.4 This condition represents an intersection between pharmacology and autoimmunity, where the therapeutic benefits of a medication need to be weighed cautiously against a severely debilitating muscular disease.
The presented case report discusses a 55-year-old woman with hypertension, dyslipidaemia, Type 2 diabetes, and obesity who developed muscle weakness and recurrent falls. The subsequent diagnostic workup revealed significantly elevated creatine kinase levels, indicating an inflammatory disorder. Following extensive investigations, she was diagnosed with anti-HMGCR-induced immune-mediated necrotising myopathy. This case emphasises the importance of considering rare neuromuscular consequences and highlights the need for comprehensive diagnostic approaches to distinguish various causes of myopathy.
CASE PRESENTATION
Patient Information and History
A 55-year-old African American woman, a clerical worker with a past medical history of hypertension, dyslipidaemia, Type 2 diabetes, and obesity, presented to the emergency department. She complained of gradually worsening weakness over the past 2 months that affected her ability to perform daily activities such as combing her hair, rising from a seated position, and climbing stairs. She initially considered her symptoms to be the result of vitamin deficiency but later sought medical attention when the symptoms worsened. Her primary care physician ordered some blood tests that showed elevated liver enzymes and creatine kinase levels (11,000 IU/L), suspected to be due to adverse effects of statins. She also reported intermittent sharp chest pains not associated with physical exertion or other symptoms such as nausea or diaphoresis. She then was referred to the emergency department for further evaluation.
Clinical Findings
On examination, she had a significantly elevated blood pressure of 223/111 mmHg and proximal muscle weakness in her lower extremities.
Timeline
As detailed in Table 1, the timeline of her case presentation highlights key events and interventions.
Diagnostic Assessment
Further evaluation of complete blood count, comprehensive metabolic panel, EKG, troponin, D-dimer, creatine kinase, B-12, and aldolase was scheduled, along with discontinuation of ezetimibe and atorvastatin till investigation results were pending (Table 2).

Table 1: Timeline.
BID: twice-daily dosage; CK: creatine kinase; EKG: electrocardiogram; EMG: electromyography; ER: emergency room; IV: intravenous; IVIG: intravenous immunoglobulin; MMF: mycophenolate mofetil.
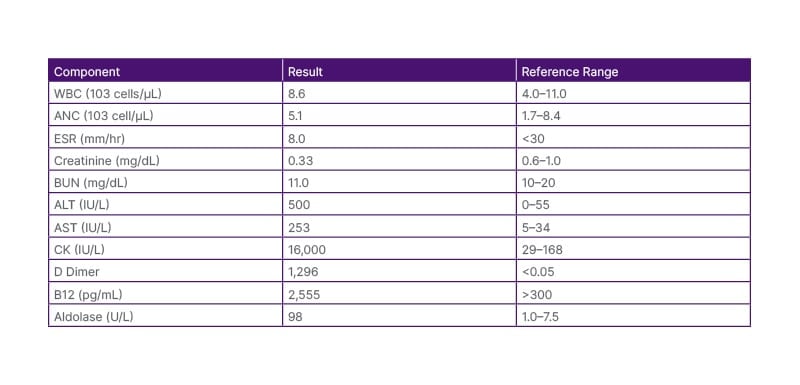
Table 2: Laboratory values upon presentation to the emergency department.
ALT: alanine aminotransferase; ANC: absolute neutrophil count; AST: aspartate aminotransferase; BUN: blood urea nitrogen; CK: creatine kinase; ESR: erythrocyte sedimentation rate; WBC: white blood cells.
The authors initially excluded the known and reversible causes of muscle weakness. The patient was admitted later to the hospital for further evaluation over the next month. Various infectious and autoimmune tests were performed, all yielding largely negative results.
A provisional diagnosis of myopathy was made, and further investigations such as nerve conduction studies, electromyography, muscle biopsy, and MRI were considered. The MRI report revealed oedema (Figure 1) in the thigh muscles, while the nerve conduction study results appeared normal. Electromyography showed irritable myopathy affecting the proximal more than the distal muscles of the upper and lower extremities.
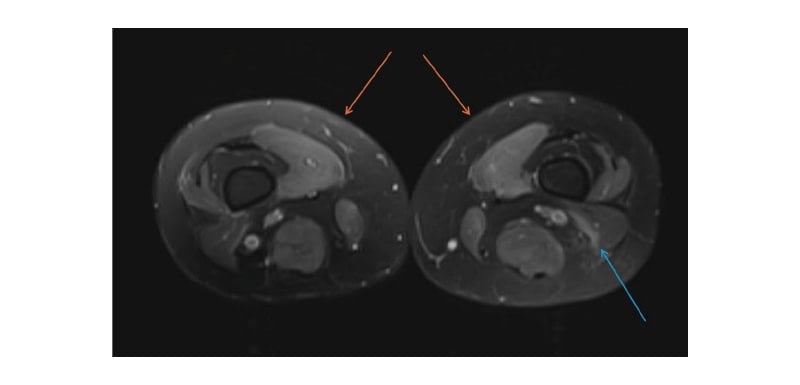
Figure 1: Axial T2-Weighted MRI of the lower extremity.
Axial T2-Weighted MRI showed symmetric hyperintensity/muscle oedema in the medial compartment (orange arrows) and posterior compartment (blue arrow).
A consistent muscle biopsy result diagnosed inflammatory necrotising myopathy. Striated muscle fibres with prominent myofibre size variation, including rounded myofibres, focal endomysial chronic inflammation (lymphocyte predominant), presence of necrotic myofibers and regenerative features, as well as patchy endomysial fibrosis and fatty replacement (perimysial>endomysial), were identified on haematoxylin and eosin (H&E) stained sections of the formalin-fixed, paraffin-embedded ‘left deltoid’ muscle biopsy material. There was no evidence of vasculitis. The collective H&E features were suggestive of inflammatory myopathy. Further antibody testing through a myopathy panel revealed positive HMGCR antibody and PM SCL 75 (polymyositis/scleroderma 75) antibody presence (Table 3).
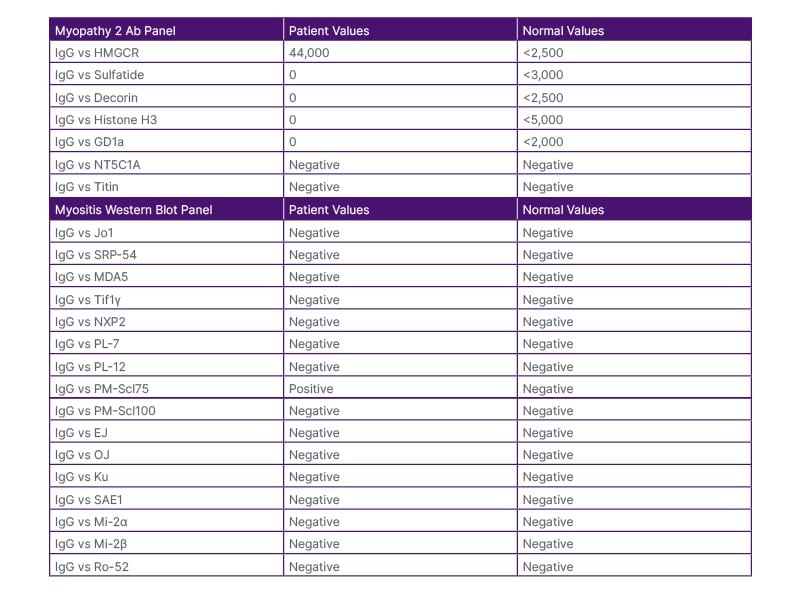
Table 3: Myositis panel results.
EJ Ab: glycyl-transfer ribonucleic acid synthetase antibody; GD1a: anti-GD1 ganglioside antibody;
HMGCR: hydroxy-3-methylglutaryl-coA reductase; Jo-1 Ab: histidyl tRNA synthetase antibody; MDA5: melanoma differentiation-associated protein-5; Mi2 alpha Ab: antibody against Mi2-alpha subunit of the nucleosome remodelling deacetylase (NURD) complex; Mi2 beta Ab: antibody against Mi2-beta subunit of the nucleosome remodelling deacetylase (NURD) complex; NT5C1A: anti-cytosolic 5′-nucleotidase 1A; NXP-2 Ab: nuclear matrix protein-2 antibody; OJ Ab: isoleucyl-tRNA synthetase antibody; PL-7 Ab: anti-threonyl-tRNA synthetase antibody; PL-12 Ab: alanyl-tRNA synthetase antibody; SAE1: anti–small ubiquitin-like modifier activating enzyme; SRP Ab: signal recognition particle antibody; TIF-1y Ab: anti-transcriptional intermediary factor 1y antibody; vs: versus.
Therapeutic Interventions and Management
The management of the authors’ patient followed a multi-dimensional and comprehensive course starting with the withdrawal of statin therapy at presentation. The treatment was initiated by aggressive immunosuppression, which included the intravenous administration of methylprednisolone at 1,000 mg/day for 3 days. This was then followed by oral prednisone at a dose of 1 mg/kg/day. Close attention was paid towards monitoring her blood glucose levels, owing to her concurrent diabetes, and her medications were adjusted accordingly, thus mandating a close collaboration with the department of endocrinology. Simultaneously, supportive care was instituted, which included calcium supplementation at 1,200 mg/day with vitamin D supplementation. Blood pressure and glycaemic control were monitored closely, and medication was adjusted appropriately throughout this period.
The patient’s creatine kinase levels gradually decreased to 9,299 U/L, 9,671 U/L, and 8,466 U/L over the course of several days following treatment. However, the levels plateaued and did not normalise. As the disease course continued to progress, treatment was escalated to include second-line agents. Intravenous immunoglobulin (IVIG) therapy was started with a loading dose of 2 g/kg, given over a period of 5 days, and thereafter dosed monthly at 1 g/kg. Due to the necessity of long-term immunosuppressive therapy, steroid-sparing agent- mycophenolate mofetil was initiated at 500 mg twice-daily dosing and titrated stepwise based on clinical response and tolerability. It included regular monitoring of complete blood count and liver function throughout this period. Due to partial response to mycophenolate mofetil, another immunosuppressive agent was considered.
Biological therapy with rituximab was instituted after proper screening for hepatitis B. Before initiating rituximab, a thorough premedication regimen was instituted with the aim of minimising infusion reactions. Induction doses of 1,000 mg were administered at Weeks 0 and 2 intravenously. The maintenance schedule was based on clinical response and B cell monitoring.
Rehabilitation played an important role in this patient’s recovery. Physical therapy intervention started with a baseline functional assessment. This was then followed by the implementation of a graduated exercise programme, progressive resistive exercises, range of motion exercises, gait training, and balance exercises with muscle strengthening. Occupational therapy centred on assessing and modifying the person’s home, teaching energy conservation techniques, and training the individual in adaptive equipment use as required.
Long-term management was based on an individualised tapering schedule of immunosuppression, keeping watch for relapse of disease. It would demand a multi-disciplinary team care involving specialists from rheumatology, neurology, endocrinology, and physical medicine and rehabilitation. Education of the patient was detailed with information regarding the understanding of the disease, warning signs of disease relapse, adherence to medications, and activity modifications appropriately. Lifestyle modification was provided in the form of exercise prescription, dietary counselling, stress management, and sleep hygiene.
Significant attention was also given to the in-place support systems. This included family education and involvement, contacting a patient support group if necessary, regular psychological assessment, and social work consultation where necessary. This holistic management approach ensured that all aspects of the condition, including both the physical and psychosocial features of the patient’s condition, were taken into account.
DISCUSSION
Statin-induced NAM is an uncommon yet significant adverse effect of statin treatment. Due to its infrequency, specific figures for its occurrence and frequency are not well established. It affects a small proportion of individuals who use statins, a frequently prescribed medication for controlling high cholesterol levels and reducing cardiovascular risk.3,5
Despite being rare, statin-induced NAM is a severe illness. It results in considerable morbidity due to severe muscle weakness and functional limitations. While the mortality directly caused by NAM is not well-defined, it significantly affects quality of life and can lead to disability. The long-lasting muscle weakness and functional limitations in patients even getting proper care and treatment emphasise the persistent nature of the illness.
The condition, predominantly but not always triggered by statins, is identified by muscle weakness in the torso area, elevated serum creatine kinase levels, and myopathic patterns detected through electromyography.4,6,7 Some studies suggest a direct relation between creatine kinase levels and muscle strength.8 Clinical symptoms, ranging from myalgia to rhabdomyolysis,9 often improve alongside decreased levels of anti-HMGCR antibodies. For instance, after treatment, undetectable antibody levels were observed in four out of seven patients.10
The pathogenesis of statin-induced NAM includes an immune-related assault on muscle fibres, marked by present antibodies against HMGCR. Statins increase the production of HMGCR in damaged muscle cells, potentially initiating an autoimmune reaction in vulnerable individuals.3,5 Statin-induced NAM is treated by stopping statins and starting immunosuppressive therapy. The initial treatment usually involves corticosteroids, sometimes combined with other immunosuppressants like methotrexate or azathioprine.6,7,11
The outlook for individuals with statin-induced necrotising autoimmune myopathy can differ. While some patients experience a positive response to treatment, as shown by the disappearance of anti-HMGCR antibodies and improved muscle strength, others may continue to experience lasting disability, as evidenced by persistent functional limitations and reduced Karnofsky Performance Status (KPS) scores.10 On average, KPS scores decreased from 89/100 before diagnosis to 68/100 at the most recent follow-up assessment.
Additional research is critical for improving the management of statin-induced NAM. Prospective clinical trials are necessary to establish standardised treatment protocols and explore the long-term outcomes of different therapeutic approaches. Investigating the mechanisms underlying persistent functional impairments despite clinical remission could provide valuable insights into improved supportive care and rehabilitation methods. Furthermore, researchers can identify genetic or other biomarkers predicting susceptibility to statin-induced NAM and can make personalised prevention strategies for patients who are on statins.
The management of anti-HMGCR immune-mediated necrotising myopathy is challenging, and standardised treatment protocols have not been established yet. Recent literature varies with the response rate to different therapeutic approaches. In a multicentre study on 100 patients with anti-HMGCR myopathy, 90% required multiple immunosuppressive agents for adequate disease control. Complete response to initial treatment with high-dose corticosteroids at 1 mg/kg/day was achieved in only 25% of cases.7,8 Second-line agents, particularly IVIG, have been promising, with one retrospective study showing improvement in 85% of patients receiving combination therapy with IVIG and corticosteroids.11 Rituximab has become an effective treatment for refractory cases, demonstrating complete or partial response in 75% of patients who failed first-line therapy.11-13 Notably, early aggressive therapy does appear to correlate with improved results, as reported by Christopher-Stine et al.,6 in that patients treated with combination immunotherapy within the first 3 months of presentation had significantly more complete remissions (68% versus 30%) than those treated later.6 Long-term follow-up data suggested by Turrin et al.10 appear to suggest that about 40% of these patients remain well at 24 months, and 20% relapse into disease upon attempts to taper their immunosuppression.4,10 Studies have also focused on the role of exercise in reducing steroid dosage and improving the quality of life in patients with multiple subtypes of idiopathic inflammatory myopathies.14 More research needs to be done specifically for statin-induced myopathies.
Patient Perspective
From the patient’s perspective, they expressed high satisfaction with the treatment and demonstrated consistent compliance, attributing their positive experience to an accurate diagnosis.
Informed consent
Informed consent was obtained from the patient prior to the commencement of the case report.
CONCLUSION
Statin-induced NAM is a rare but significant adverse effect of statin therapy, characterised by severe muscle weakness, elevated serum creatine kinase levels, and myopathic changes in electromyography. Despite its infrequency, the impact on affected individuals is substantial, often leading to considerable morbidity and functional limitations. Due to its autoimmune nature, marked by anti-HMGCR antibodies, there is a need for careful monitoring and management of patients on statins.
The standard treatment involves discontinuing statin use and initiating immunosuppressive therapy, primarily with corticosteroids, often supplemented with additional immunosuppressants like methotrexate or azathioprine. While some patients respond well to treatment, achieving remission and improvement in muscle strength, others continue to experience persistent disability and reduced quality of life, as reflected in decreased KPS scores.
THE WAY FORWARD
Further research is essential to enhance the management and understanding of statin-induced NAM. Prospective clinical trials are crucial for developing standardised treatment protocols and for evaluating the long-term efficacy and safety of different therapeutic regimens. Additionally, investigating the underlying mechanisms of apparent clinical remission with persistent functional impairments could lead to better supportive care and rehabilitation strategies.
Moreover, identifying genetic or other biomarkers that predict susceptibility to statin-induced NAM is critical. Such advancements could enable personalised prevention strategies, ensuring that patients at higher risk can be monitored more closely or offered alternative treatments. Ultimately, improving the management of statin-induced NAM will mitigate the adverse effects on patients’ quality of life and enhance the overall safety profile of statin therapy.