
Abstract
Neratinib was developed as an irreversible catalytic inhibitor of ERBB2, which also acts to inhibit ERBB1 and ERBB4. Neratinib is U.S. Food and Drug Administration (FDA)-approved as a neo-adjuvant therapy for use in HER2+ breast cancer. More recently, chemical biology analyses and the authors’ own bench work have demonstrated that neratinib has additional targets, which open up the possibility of using the drug in cell types that either lack ERBB receptor family expression or who rely on survival signalling downstream of growth factor receptors. Neratinib rapidly disrupted mutant RAS nanoclustering, which was followed by mutant rat sarcoma virus proteins translocating via LC3-associated phagocytosis into the cytosol where they were degraded by macroautophagy. Neratinib catalytically inhibited the MAP4K mammalian STE20-like protein kinase 4 and also caused its degradation via macroautophagy. This resulted in ezrin dephosphorylation and the plasma membrane becoming flaccid. Neratinib disrupted the nanoclustering of RAC1, which was associated with dephosphorylation of PAK1 and Merlin, and with increased phosphorylation of the Merlin binding partners large tumour suppressor kinase 1/2, YAP, and TAZ. YAP and TAZ exited the nucleus. Neratinib retained its anti-tumour efficacy against NSCLC cells made resistant to either afatinib or to osimertinib. Collectively, these findings argue that the possibilities for the further development of neratinib as cancer therapeutic in malignancies that do not express or over-express members of the ERBB receptor family are potentially wide-ranging.
Key Points
1. Neratinib was developed as an neoadjuvant irreversible inhibitor of catalytic ERBB2 for use in patients with HER2+ breast cancer.
2. Chemical biology studies have identified additional targets for netatinib, through the disruption of mutant RAS nanoclustering and its other anti-tumour effects.
3. Although associated with gastrointestinal toxicity, alterations to the administration route of neratinib has shown a relatively benign safety profile.
THE ‘NEW’ BIOLOGY OF NERATINIB
Neratinib (HKI-272) was developed as an irreversible inhibitor of ERBB2 (HER2) catalytic activity, which was also shown to inhibit ERBB1 (the EGF receptor) and ERBB4.1,2 It gained U.S. Food and Drug Administration (FDA) approval in the USA in 2017 and by the European Union (EU) in 2018, for use in patients with HER2+ breast cancer in combination with the 5-fluorouracil analogue capecitabine.3,4 Neratinib was originally developed by Wyeth and Pfizer (New York, USA), and then licensed to Puma Biotechnology (Los Angeles, California, USA) in 2011.5 All small molecule therapeutic agents have recognised on-target and off-target effects, although such was the focus of neratinib development on HER2+ tumours that the off-target biology of the drug, in vitro and in patients, was not thoroughly investigated. In 2011 Davis et al.6 published an extensive chemical biology screening study of over 40 kinase inhibitors, that included neratinib.6 In their analyses, Davis et al.6 argued that neratinib was an inhibitor of multiple MAP4K serine or threonine kinases including mammalian STE20-like protein kinase 4 (MST4). The most notable negative sequela of neratinib during its clinical development was diarrhoea and it is known that MST4 plays an essential role in the development of the gastrointestinal brush border.7 These data strongly argue that MST4 is a validated in vivo ‘off-target’ for neratinib.
The authors’ studies over the past 4 years examining neratinib have been performed in afatinib-resistant and osimertinib-resistant non-small cell lung cancer cells that express mutant active ERBB1 proteins, and in pancreatic, colorectal, cutaneous melanoma, and uveal melanoma cancer cells that express mutant KRAS, mutant NRAS, and mutant Ga proteins.8-16 Mutant rat sarcoma virus (Ras) proteins and mutant Ga proteins lack guanosine triphosphate (GTP)ase activity and cannot hydrolyse GTP back to guanosine diphosphate; these are hence considered ‘constitutively active’. In the authors’ studies, they made two unexpected discoveries. First, was that neratinib was not only an irreversible inhibitor of the ERBB receptors but that it also caused receptor internalisation and the subsequent degradation of the receptors via LC3-associated phagocytosis and macroautophagy. Other growth factor receptors which can associate with super-complexes of ERBB receptors in the plasma membrane such as c-MET and PDGFRb, could, also in a cell-type-dependent fashion, be internalised and degraded. Second, based on the authors’ receptor internalisation data, the authors hypothesised that neratinib would also cause the internalisation of plasma membrane-localised GTP binding proteins, i.e., Ras proteins and heterotrimeric Ga proteins. In both pancreatic and colon cancer cells neratinib caused endogenous mutant K-Ras proteins to localise in vesicles liminal to the plasma membrane; vesicles that co-stained for the macroautophagy protein Beclin1. Over a 6-hour time course, the expression levels of the mutant K-Ras and mutant N-Ras proteins in cutaneous melanoma, pancreatic, and colon cancer cells fell by approximately 30%. In patient-derived xenografts (PDX) uveal melanoma lines, neratinib as a single agent within 6 hours significantly reduced the protein levels of Gaq and Ga11 by approximately 50%. In cells transiently transfected with plasmids to express green fluorescent protein (GFP) and red fluorescent protein (RFP) tagged forms of K-Ras G12V, neratinib rapidly caused the formation of GFP+ RFP+ (yellow) vesicles that subsequently only fluoresced RFP+, and at a lower fluorescence intensity. This data is indicative that the Ras proteins were initially localised in autophagosomes and that autophagic flux had occurred with the GFP+ signal being quenched in the acidic autolysosome, and with degradation of the Ras fusion proteins.
Signalling by Ras proteins out of the plasma membrane and into the cytosol requires that not only are they in their GTP-bound state but also that they form loosely binding nanoclusters of 5–6 proteins, each with a radius of approximately 9 nm and with a half-life of less than one second.17-23 Based on the concept of rapidly accelerated gibrosarcoma (RAF) protein dimerisation and cross-talk between RAF proteins, e.g., by B-RAF and RAF-1 as homo- and hetero-dimers, to mediate their activation and that of the downstream extracellular signal-regulated kinase 1/2 (ERK1/2) pathway, the concept that Ras proteins also having to form nanoclusters to foster RAF–RAF interactions for effective signalling off the plasma membrane is logical.24 Using immuno-gold electron microscopy, the authors observed that the initial impact of neratinib upon Ras biology was to rapidly disrupt Ras nanoclustering in the plasma membrane that was temporally followed by Ras mislocalisation away from the plasma membrane to the intracellular site(s). Stable transfection of cells to express ERBB1 neither altered the effect of neratinib on mutant KRAS nanoclustering nor in the mutant KRAS subsequently mislocating from the plasma membrane into the cytosol. In haematopoietic cells that do not express ERBB receptors, neratinib caused Ras proteins to mislocate from the membrane to the cytosol. Clearly, for its Ras biology, receptor binding was not required.
The small GTP binding protein RAC1 also forms nanoclusters in the plasma membrane as a pre-requisite for signalling into the cell, and neratinib disrupted RAC1 nanoclustering, though unlike RAS proteins, this did not result in RAC1 mislocalisation to the cytosol. Reduced RAC1 nanoclustering was associated with the dephosphorylation of its direct interacting partner, the kinase PAK1, and of the PAK1 substrate, Merlin.25 In parallel to these events, the plasma membrane of neratinib-treated cells became flaccid and the cytoskeletal target of MST4, Ezrin, which regulates plasma membrane rigidity and its interaction with the cytoskeleton, was dephosphorylated (Figure 1).26

Figure 1: Amino acid sequence alignment comparing KRAS and RAC1.
Neratinib acts as an irreversible inhibitor of ERBB family receptors by covalently linking to a cysteine residue in their active sites.27 All Ras proteins and RAC1 only share a single conserved cysteine amino acid at position 80/81, and, at present, it is not known whether neratinib covalently attaches via this cysteine residue to Ras/RAC proteins to disrupt both their nanoclustering and their signalling off the plasma membrane and into the cell. To initially address this issue, the authors transfected cells to express KRASG12V-GFP. The authors treated cells with neratinib maleate and lysed the cells 5 min, 10 min, and 30 min after drug exposure. The authors immunoprecipitated KRASG12V–GFP using the GFP tag. The authors then ran the precipitate on a sodium dodecyl-sulfate polyacrylamide gel electrophoresis and blotted for RAF-1. Within 5 min, the amount of RAF-1 that co-precipitated with KRASG12V-GFP was significantly reduced, and the amount of RAF-1 co-precipitating further declined after 30 min. This strongly argues that neratinib was reducing the ability of RAF-1 to associate with KRASG12V (unpublished findings). If validated by additional experimentation, this would make neratinib, at present, a unique drug, and a claimant to be ‘the holy grail’ drug that can directly inhibit mutant Ras proteins. Even so, neratinib can perhaps only be considered as a ‘prototype’ Ras inhibitory drug, and additional chemical biology work will be required to design and develop a truly potent Ras inhibitor. However, direct covalent association with Ras and RAC1 proteins may not be the only possible mechanism at play because in the authors’ studies they also discovered that neratinib significantly reduced the level of phosphatidyl serine in the plasma membrane and increased the levels of cholesterol. Increased membrane cholesterol is congruent with greater membrane fluidity, as the authors had previously observed by light microscopy. And increased membrane fluidity is known to independently disrupt mutant RAS signalling. How neratinib alters phosphatidyl serine and cholesterol levels will also require additional studies.
Figure 2 is the collation of the authors’ signal transduction data derived from findings in multiple tumour cell lines expressing mutant KRAS or NRAS proteins and treated with neratinib and pemetrexed. Neratinib rapidly disrupts RAS signalling concomitant with reduced phosphorylation (activity) of RAF-1 (S338), MEK1/2 (S218/S222), and ERK1/2 (T183/Y185), as well as, in the PI3K pathway, of AKT (T308 and S473), mTORC1 (S2448), mTORC2 (S2481), and p70 S6K (T389 and S424). The authors presume these broad effects are due to both reduced RAS signalling per se and reduced signalling by ERBB family and other receptor tyrosine kinases. Published data shows that after 8 hours of neratinib exposure, significant amounts of Ras protein have been degraded.Neratinib as a single agent causes activation of ATM (S1981) by the generation of reactive oxygen species, with ATM-dependent phosphorylation of AMPKαT172. Pemetrexed as a single agent also activates ATM via DNA damage, and via its inhibition of AICART it also enhances the levels of the nucleoside ZMP, which like AMP, allosterically activates the AMPK. Thus, neratinib and pemetrexed interact to cause ‘full’ AMPK activation and this event co-operates with inactivation of AKT due to the loss of RTKs and Ras to cause the inactivation of mTORC1 and mTORC2. Full AMPK activation enhances ULK1 S317 phosphorylation and mTORC1 inactivation lowers ULK1 S757 phosphorylation collectively resulting in ‘full’ activation of ULK1. Fully activated ULK1 phosphorylates ATG13 S318, the gatekeeping phosphorylation event required to trigger autophagosome formation, ultimately leading to greater autophagic flux and autophagy-dependent tumour cell killing. In parallel to these events, neratinib causes inactivation and degradation of the MAP4K MST4, which ultimately leads to enhanced phosphorylation of large tumour suppressor kinase 1/2, YAP, and TAZ with both co-transcription factors leaving the nucleus (i.e., their inactivation). Loss of YAP/TAZ function reduces the synthesis of multiple autocrine growth factors and is associated with a reduced ability of tumour cells to migrate and metastasise. Neratinib degrades the chaperone GRP78 via autophagy, which results in the activation of PKR-like endoplasmic reticulum kinase, which phosphorylates and inactivates eIF2a S51 (i.e., an ‘ER stress response’). The actions of phosphorylated eIF2a result in both reduced expression of cytoprotective proteins with short half-lives such as FLIP-s, MCL1, and BCL-XL, and with increased expression of the autophagy regulatory proteins Beclin1 and ATG5, which, respectively, enhances death receptor signalling and mitochondrial dysfunction and sustains toxic autophagosome formation. All of these events act to enhance drug-induced tumour cell killing.
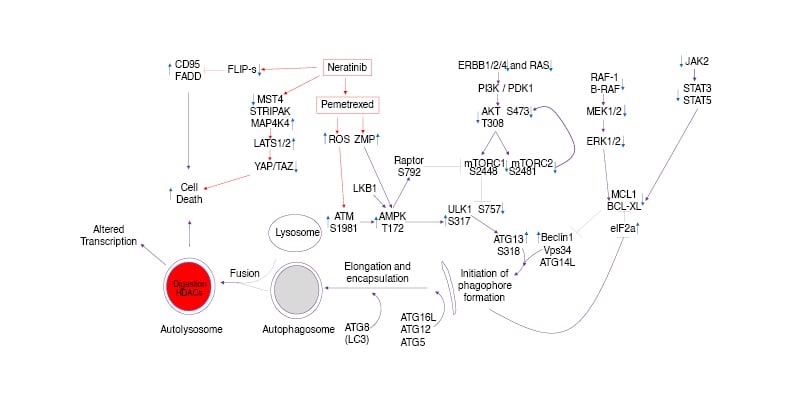
Figure 2: Signalling pathways whose activities are directly and indirectly altered when non-small cell lung carcinoma cells are exposed to neratinib and the anti-metabolite pemetrexed.
AMPK: AMP-dependent protein kinase; ER: endoplasmic reticulum; ERK: extracellular regulated kinase; FADD: FAS-associated death domain protein; HDAC: histone deacetylase; LAP: LC3-associated phagocytosis; LATS: large tumour suppressor kinase; MAPK: mitogen activated protein kinase; MST4: mammalian STE20-like protein kinase 4; mTOR: mammalian target of rapamycin; PI3K: phosphatidyl inositol 3 kinase; PTEN: phosphatase and tensin homologue; ROS: reactive oxygen species; STRIPAK: striatin-interactingphosphatase and kinase; STAT: signal transducers and activators of transcription.
Neratinib, acting at the level of growth factor receptors and plasma membrane GTP binding proteins, would not be considered to have any activity in tumour cells that express intracellular oncogenes such as cutaneous melanomas expressing B-RAF (V600E). The treatment of tumours expressing this oncogene has been revolutionised by the development of small molecule kinase inhibitors that selectively inhibit the mutant forms of B-RAF such as dabrafenib, and the combination of these drugs with MEK1/2/5 inhibitors such as trametinib, which prevents compensatory survival signalling from RAF-1 causing reactivation of the ERK1/2 pathway.28,29 It has been recognised for over a decade that tumour cells expressing wild-type K-Ras and NRAS proteins can behave in a de facto manner very similar to cells expressing mutant Ras proteins due to the over-expression of paracrine factors and the constitutive activation of multiple upstream growth factor receptors.30,31 As such, the authors hypothesised that neratinib, by causing growth factor receptor and Ras protein degradation, and possibly also by causing the degradation of RAF-1/B-RAF all via macroautophagy, would have anti-tumour efficacy in cutaneous melanoma cells expressing mutant B-Raf proteins.
The authors initial studies compared the anti-tumour efficacy of neratinib in vitro alongside that of the established trametinib/dabrafenib combination in PDX models of cutaneous melanoma expressing the B-RAF (V600E) oncogene. The authors’ first observation of note was that after 24 hours of drug exposure, low nanomolar concentrations of neratinib as a single agent, below its safe plasma C max, caused similar to greater amounts of cell death when compared with the killing effect caused by the trametinib/dabrafenib drug combination. Drug naïve MEL4 cells expressed higher levels of B-RAF and RAF-1 when compared with vemurafenib-resistant MEL2 cells. Neratinib reduced B-RAF (V600E) and RAF-1 expression in drug naïve MEL4 cells but not in vemurafenib-resistant MEL2 cells. The observed reduction of B-RAF and RAF-1 effectively normalised protein levels between the two PDX isolates, and similar levels of killing by neratinib were observed in both the MEL2 and MEL4 cells. Although neratinib could be causing the degradation of wild-type RAF-1 and wild-type B-RAF located at the plasma membrane because they are associated with Ras proteins, the authors also are cognisant that neratinib is a potent stimulator of autophagosome formation, and via macroautophagy, causes the degradation of cytosolic HSP90 and HDAC6. HSP90 is a key chaperone protein regulating the stability of both RAF-1 and B-RAF. HDAC6 specifically de-acetylates HSP90, enhancing its chaperoning activity. Thus, independently of plasma membrane association, neratinib can act to destabilise cytosolic RAF-1 and B-RAF proteins.
Neratinib, as with ERBB receptors, not only catalytically inhibits the serine/threonine kinase MST4, but in addition, causes its autophagic degradation. MST4 is one component of striatin-interacting phosphatase and kinase (STRIPAK) complexes.32,33 These complexes are plasma membrane liminal supra-molecular assemblies of proteins with components including: striatins 3 and 4, MST4, MAP4K4, ezrin, radixin, and Moesin proteins, sarcolemmal membrane-associated protein, and protein serine/threonine phosphatase 2A. The authors published studies in other tumour cell types have demonstrated that neratinib: selectively activated MAP4K4 but not MAP4K1; and inactivated MST4, caused MST4 degradation and Ezrin dephosphorylation.
Both events will rapidly alter the composition of the STRIPAK complexes, and one downstream event associated with this is the activation of large tumour suppressor kinase 1 and 2. This, in turn, results in the phosphorylation of YAP and TAZ, co-transcription factor effectors for the Hippo pathway, and causes YAP and TAZ to exit the nucleus. In cutaneous melanoma cells, the authors also found that neratinib increased the phosphorylation of YAP and TAZ at multiple sites and caused their nuclear exit.11-16 YAP and TAZ are de facto oncogenes, which can substitute for RAS mutations and, in particular, they are known to promote the metastatic spread of tumour cells.34,35 Hence, the authors’ data working with neratinib has argued, not only in cutaneous melanoma cells but also in other solid and liquid tumour cell types, that regardless of drug-resistance or the lack of ERBB family receptor expression, the multi-factorial targeting of neratinib against multiple protein kinases regulating multiple signalling pathways demonstrates its broad potential utility in the treatment of cancer.
NERATINIB AND FUTURE CHALLENGES
Based on the authors’ neratinib research, there are two open Phase I clinical trials, NCT0391929236 and NCT04502602.37
Up until its regulatory approvals in 2017 and 2018, neratinib had a poor reputation in the clinic during its development due to it causing Grade 3/4 diarrhoea in the majority of patients. This issue was resolved by prophylactic use prior to, and the continuing use of, loperamide-based anti-diarrhoeal drugs during therapy. The fact remains, however, that gastrointestinal toxicity is a limiting factor in the deployment of neratinib not only against HER2+ breast cancer but also against all other tumour cell types, particularly those expressing mutant Tas or G α proteins. Based on data obtained during its development and its present use in the clinic, neratinib appears to have a relatively benign safety profile once in the plasma against the cells that form the hematocrit and line blood vessels. This suggests that the development of an intravenous neratinib formulation, which would result in a safe plasma C max values of greater than the present approximately 150 nM for oral administration could dramatically enhance the therapeutic utility of the drug against other tumour types.
In addition to being an anti-cancer drug, it also may be possible to develop neratinib as a novel therapeutic for Alzheimer’s Disease (AD).38 Aberrant expression and denaturation of Tau, amyloid-β, and TDP-43 can lead to cell death and is a major component of AD pathologies. AD neurons exhibit a reduced ability to form autophagosomes and degrade proteins via autophagy. Neratinib via macroautophagy reduced chaperone levels and the expression of Tau, Tau 301L, APP, APP692, APP715, SOD1 G93A and TDP-43 in cancer cells, neuronal cells, and microglia. Further work will be required to determine in vivo whether neratinib slows down AD progression.






