Abstract
Background: Immunoglobulin D (IgD) myeloma is an uncommon subtype of multiple myeloma that accounts for 1–2% of all myeloma cases. IgD myeloma is often associated with a high incidence of renal insufficiency, hypercalcaemia, extraosseous disease, amyloidosis, and Bence–Jones proteinuria.
Materials and Methods: The authors discuss the clinical presentation and treatment results of 12 cases of IgD myeloma, treated at a single centre.
Results: The study included nine males and three females, with a median age of 48.5 years. All had lytic bone lesions, eight had anaemia, five had renal impairment, three had hypercalcaemia, three had features of amyloidosis, and one had polyneuropathy, organomegaly, endocrinopathy, M-protein, and skin changes syndrome. On immunofixation electrophoresis, 10 patients had IgD λ subtype, and two had IgD κ. Urine Bence–Jones protein was positive for five patients. Eleven patients received bortezomib-based chemotherapy. Three patients achieved complete response, two had a very good partial response, two achieved partial response,three progressed, and one discontinued treatment. At 2 years, six patients were alive (50%). Median overall survival of the series was 21 months (range: 1–80 months).
Conclusion: IgD myeloma is an uncommon subtype of myeloma associated with a high incidence of hypercalcaemia, renal insufficiency, extraosseous lesions, amyloidosis, and Bence–Jones proteinuria. IgD λ disease is more frequent, unlike other subtypes of IgD myeloma. Management is similar to other myeloma types, but with poorer outcomes.
Key Points
1. Immunoglobulin D (IgD) myeloma is a rare subtype of multiple myeloma, which accounts for less than 2% of all myelomas. It is often associated with a high incidence of renal failure, hypercalcaemia, extraosseous disease, amyloidosis, Bence–Jones proteinuria, and a small or absent M spike. Patients with IgD myeloma have a poor outcome compared to other subtypes.2. This study describes the clinical presentations, laboratory findings, treatment received, and outcome of 12 patients with IgD multiple myeloma, treated in a tertiary cancer centre. A literature review of clinical characteristics, and treatment outcome of studies in IgD myeloma published in literature, is also included.
3. IgD myeloma affects relatively younger males, and presents in advanced International Staging System (ISS) stage. IgD λ disease is more frequent, unlike other subtypes of myeloma. Since IgD levels are generally very low in the serum, it may escape detection. Treatment of IgD myeloma is similar to other subtypes of myeloma. Bortezomib-based chemotherapy is an effective regimen.
INTRODUCTION
Multiple myeloma (MM) is a disease characterised by the clonal expansion of malignant plasma cells in the marrow, leading to anaemia, hypercalcaemia, bone lesions, and renal dysfunction. MM accounts for approximately 10% of haematologic malignancies, and 1% of all cancers.1 MM can be of various types, involving different isotypes of immunoglobin (Ig) heavy chains and light chains. IgG, IgA, and light chain myelomas are the most prevalent, comprising 52%, 21%, and 16% of MM cases, respectively. Together, they constitute 90% of all MM cases. Other MM types are IgD, IgE, IgM, and non-secretory types.2,3
IgD myeloma is a rare subtype of MM, accounting for <2% of all myelomas.4-6 It is often associated with a high incidence of renal failure, hypercalcaemia, extraosseous disease, amyloidosis, and Bence–Jones proteinuria.7-9
As the secretion of IgD is low in the serum, it is difficult to confirm the presence of monoclonal protein IgD by serum protein electrophoresis or immunofixation electrophoresis.10 Patients with IgD myeloma have a poor outcome compared to other subtypes, with a median survival of 13–21 months.11,12 Despite its aggressive course, recent advances in the treatment of MM have improved patient outcomes in IgD myeloma.
In this report, the authors present the results of a retrospective analysis of 12 cases of IgD myeloma, treated at a tertiary cancer centre.
OBJECTIVES
The objective was to study the clinical presentations, treatment, and survival of patients with IgD myeloma treated in a single centre.
MATERIALS AND METHODS
This is a retrospective study conducted at a tertiary cancer centre, in the Department of Medical Oncology, Regional Cancer Centre, Thiruvananthapuram, India, on 12 patients diagnosed with IgD myeloma during a 5-year period, from January 2015–December 2019. The details of clinical presentation, diagnosis, treatment, and survival were noted from medical records. All procedures performed in this study were in accordance with the ethical standards of the institutional and/or national research committee, and with the 1964 Helsinki Declaration and its later amendments, or comparable ethical standards.
TREATMENT PROTOCOL
The standard treatment for MM included systemic chemotherapy with the proteasome inhibitor bortezomib, along with other drugs, or a combination of lenalidomide and dexamethasone, as induction. This was followed by bortezomib or lenalidomide maintenance chemotherapy. The patient response was assessed at regular intervals during induction and maintenance therapy. Radiotherapy was considered for patients with spinal cord compression, and for pain relief.
STATISTICAL METHODS
The baseline patient characteristics, diagnosis, treatment details, and response assessment were analysed using descriptive statistics, including frequency, percentage, median, range, and mean. Overall survival (OS) was calculated from the date of initial diagnosis to the date of death from any cause, or last follow-up visit. Progression-free survival (PFS) was calculated from the initiation of chemotherapy until disease progression.
RESULTS
Among 1,240 patients with MM seen between 2015–2019 at the authors’ centre, 12 patients were diagnosed with IgD myeloma, with an incidence of 0.96%. The median age was 48.5 years (range: 34.0–67.0 years). There were nine males and three females. Eight patients presented with bone pain, and three patients presented with features of amyloid deposits. One patient had features of polyneuropathy, organomegaly, endocrinopathy, M-protein, and skin changes (POEMS) syndrome. Among the 12 cases, all had lytic bone lesions, eight had anaemia (haemoglobin <10 g/dL), five had renal impairment (serum creatinine >2 mg/dL), and three had hypercalcaemia (>11.5 mg/dL). The median duration of symptoms was 8 weeks (range: 4–24 weeks). The baseline characteristics of all patients are summarised in Table 1.
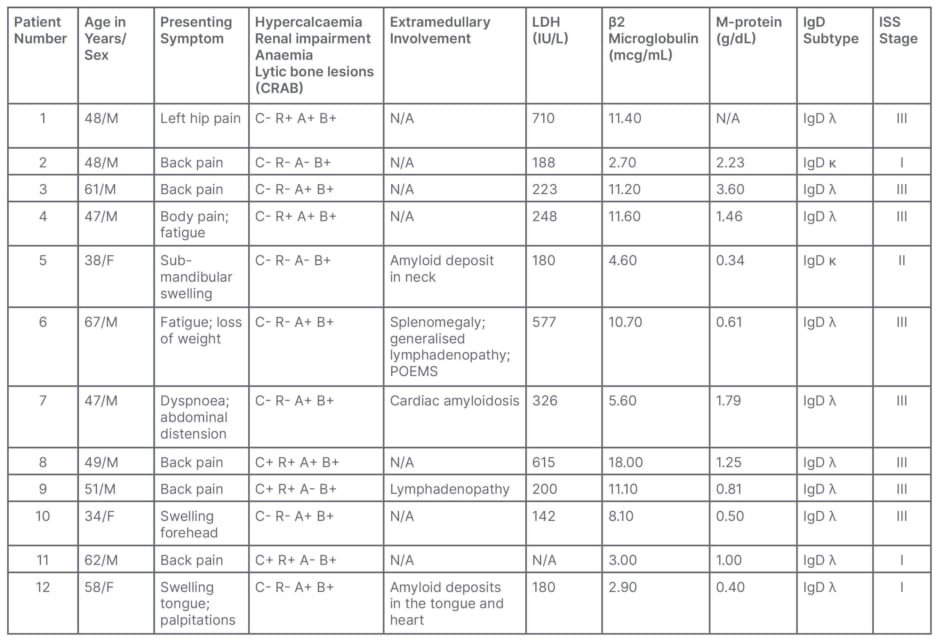
Table 1: Summary of clinical presentation and diagnosis of 12 patients with immunoglobin D myeloma.
ISS: international staging system; LDH: lactate dehydrogenase; N/A: not applicable; POEMS: polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy and skin changes.
Mean haemoglobin was 10.13 g/dL (standard deviation [SD]: 2.12 g/dL). Two patients had thrombocytopenia. Mean albumin was 4.05 g/dL (SD: 0.47 g/dL), and β2 microglobulin was 8.40 mcg/mL (SD: 4.72 mcg/mL). The albumin/globulin ratio was reversed in four patients. According to the International Staging System (ISS), three patients had Stage I MM, one patient had Stage II, and eight patients had Stage III. Median serum lactate dehydrogenase was 223 IU/L (range: 142–710 IU/L [normal range: 120–246 IU/L]). Six patients had a prominent M-band of >1 g/dL, with a median M-spike of 1 g/dL (range: 0.34–3.6 g/dL). On immunofixation electrophoresis, 10 patients had IgD λ subtype, and two had IgD κ. On free light chain assay, the median serum κ was 23.5 mg/L (range: 8.8–5,395.0 mg/L) and serum λ was 1,765.1 mg/L (range: 11.8–6,935.0 mg/L). The ratio of involved to uninvolved free light chain was >100 in seven patients. The median 24-hour urine protein was 2,000 mg (range 147–4,095 mg). Urine Bence–Jones protein was positive in five patients. Urine electrophoresis was available in 10 patients, of whom eight showed a band in the γ-globulin region. Cytogenetics risk stratification was not done, since the facility was not available in the authors’ centre.
Six patients received induction with bortezomib, lenalidomide, and dexamethasone; two patients with bortezomib, cyclophosphamide, and dexamethasone; and three with bortezomib and dexamethasone. One patient died before treatment initiation. Two patients received radiation at a dose of 20 Gy/5# for spinal cord compression, and four patients received palliative radiation at a dose of 8 Gy/1# to symptomatic bone lesions. Response assessment was done according to the International Myeloma Working Group (IMWG) response assessment criteria. Complete response was achieved in three patients, two patients had very good partial response (VgPR), two achieved partial response, and three progressed. In one case, response could not be assessed, since the patient defaulted after the first cycle. Patients who achieved at least VgPR received maintenance with bortezomib (n=4) and lenalidomide (n=1). Three patients relapsed during maintenance. All patients who relapsed or progressed received salvage chemotherapy with carfilzomib, pomalidomide, lenalidomide, or thalidomide-based regimens. Following salvage chemotherapy, one patient achieved complete response, one achieved VgPR, and all others had persistent disease. At 2 years, six patients were alive (50%). Median OS of the series was 21 months (range: 1–80 months; Table 2).
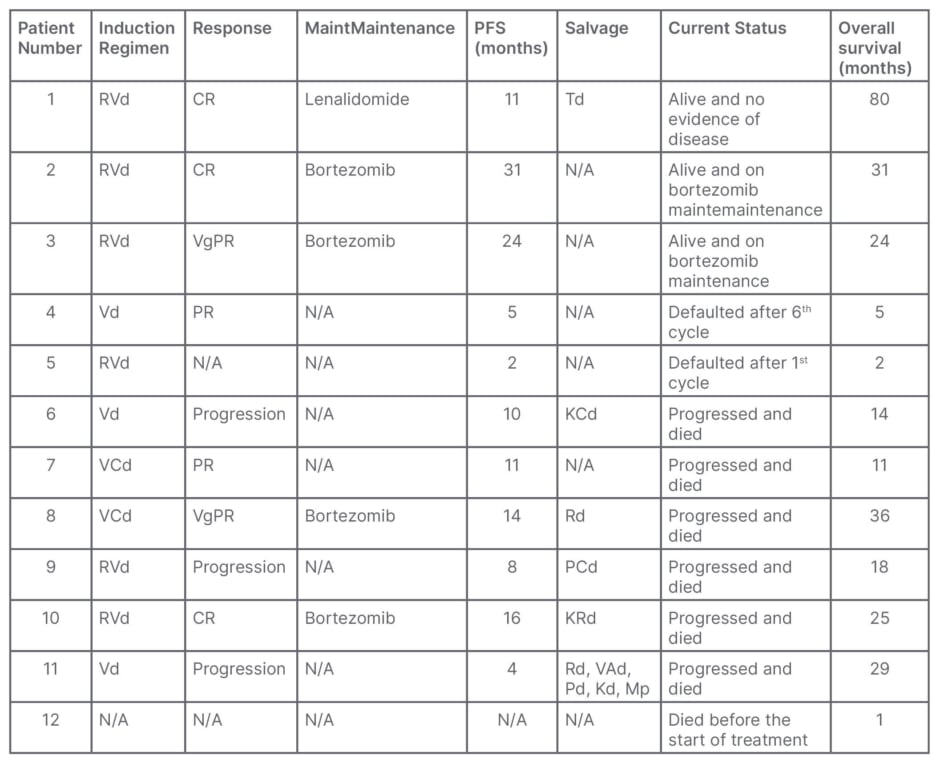
Table 2: Summary of treatment details of 12 patients with immunoglobin D myeloma.
CR: complete response; KCd: carfilzomib+cyclophosphamide+dexamethasone; Kd: carfilzomib+dexamethasone; KRd: carfilzomib+lenalidomide+dexamethasone; Mp: melphalan+prednisolone; N/A: not applicable; PCd: pomalidomide+cyclophosphamide+dexamethasone; Pd: pomalidomide+dexamethasone; PFS: progression-free survival; PR: partial response; Rd: lenalidomide+dexamethasone; RVd: nortezomib+lenalidomide+dexamethasone; Td: thalidomide+dexamethasone; VAd: vincristine+adriamycin+dexamethasone; VCd: bortezomib+cyclophosphamide+dexamethasone; Vd: bortezomib+dexamethasone; VgPR: very good partial response.
DISCUSSION
IgD myeloma was first described by Rowe and Fahey in 1965.13 Since then, studies have reported a prevalence of IgD myeloma of 1–2% among all myeloma cases.2,7,14,15 In patients <40 years, the incidence of IgD myeloma is 6%.16 The incidence in the present study was 0.96%. IgD myeloma affects relatively younger males, with an average age of 59 years (54–65 years).3,11,17 The median age in the present study was 48.5 years, which is younger than previously reported. In a recent study, IgD myeloma accounted for 4.3% of all myeloma cases, with a median age of 57.5%.18 Given the rarity of the disease, large studies on IgD myeloma are limited. Selene et al.19 conducted a systematic review of the clinical presentation, management, and prognostic factors of 166 patients with IgD MM.
The clinical features of IgD myeloma are similar to other types of myelomas. However, IgD MM has been found to involve relatively younger patients, and is characterised by extramedullary disease, osteolytic bone lesions, amyloidosis, hypercalcaemia, renal failure, Bence–Jones proteinuria, and a small or absent M-spike.3,7,14,15,20 These patients usually present in the advanced ISS stage as well.
Renal failure and Bence–Jones proteinuria are significantly higher with IgD myeloma compared to other subtypes, with renal failure present in 20–40% of patients at the time of diagnosis.11,21 Renal injury may be caused by light chain cast nephropathy, or due to direct toxicity by intracellular crystals.22,23 A recent study on risk factors of renal damage and prognosis in IgD myeloma reported that 55.3% of patients with IgD myeloma had renal damage as a complication, and that β2 microglobulin was an independent risk factor for renal damage. The median OS of the renal impairment group was significantly shorter than the non-renal impairment group.24 Bence–Jones proteinuria was reported in about 71% of IgD myeloma cases, in contrast to 35% of IgG, and 20% of IgA myeloma.3
Extramedullary disease is seen in 19–63% of patients with MM, the commonly involved sites being the chest wall, respiratory tract, gastrointestinal tract, skin, lymph nodes, and, rarely, testes.19,25 Amyloid light chain amyloidosis mostly affecting the heart is a common complication of IgD myeloma, seen in 19% of cases.3 In the present study, among 12 patients, three had amyloidosis, three had hypercalcaemia, and five had renal impairment.
In a recent study on the clinical features and prognosis of IgD myeloma, the first clinical manifestations were fatigue, bone pain, renal impairment, anaemia in 70.0% of cases, bone lesions in 60.0% of cases, renal impairment in 40.0% of cases, and hypercalcaemia in 51.4% of cases. 1q21 amplification was noted in 50% of cases, extramedullary disease occurred in 20% of cases, and 90% of patients had λ light chain disease.18
IgD-secreting plasma cells originate from somatic hypermutation of the IgD region of germinal centre B cells.19,25 In contrast to other subtypes of myeloma, there is a predominance of λ light chain in IgD myeloma (70–90%), compared to κ light chain (3–4%).26,27 An increased survival has also been reported in patients with λ subtype.5 In the present study, 80% of the light chain was λ. Recently, detection of cytoplasmic IgD by flow cytometry was identified as a sensitive way to diagnose IgD myeloma.28
Cytogenetic changes have an important role in the pathogenesis of myeloma. High risk chromosomal changes in IgD myeloma are reported to range from 33–53%.17 Complex karyotype has been reported in 21% of patients, of which 90% had TP53 gene deletion or 1q21 amplification, which is considered to be one of the poor prognostic factors in IgD myeloma.29,30 IgD levels in serum are generally very low, and may escape detection, leading to the misdiagnosis of IgD myeloma as non-secretory myeloma.19 The role of N-glycan biomarkers in the diagnosis and prognosis of IgD myeloma has also been studied.31 Egan et al.32 reported on interphase fluorescence in situ hybridisation from 29 cases of IgD myeloma. They showed a decreased OS in patients aged ≥70 years, and better outcomes in those with κ light chain restriction. Patients who were CD56-positive had longer survival than those lacking CD56.32
Agbuduwe et al.33 compared the clinical characteristics and outcomes of patients with IgD myeloma from UK Phase III clinical trials, before 2002 and after 2002. IgD myeloma comprised 1.6% and 1.2% of the two groups, respectively. It was associated with male predominance, low level of paraproteins, and λ light chains. The frequency of high-risk cytogenetics was similar compared to other types. The recent trials showed a higher response rate and median OS compared to those prior to 2002.33
Liu et al.34 carried out a multicentre retrospective study to evaluate the prevalence, clinical features, and prognosis of IgD myeloma, and to develop and validate a prognostic model, including 356 patients with IgD myeloma from 14 centres in the Asian Myeloma Network (AMN). IgD myeloma represented 2.0–8.8% of all patients with myeloma. The clinical characteristics of IgD myeloma were compared with 712 non-IgD myeloma patients. IgD myeloma was more common in males, younger patients, and patients who had advanced revised-ISS Stage III, hypercalcaemia, elevated creatinine levels, and elevated lactate dehydrogenase. A total of 40.6% of patients had abnormal karyotypes, and the frequency of chromosomal translocation t(11;14) was significantly higher than in non-IgD subtypes. However, the cytogenetic abnormalities did not have an impact on survival.34
Treatment of IgD myeloma is similar to other subtypes of myeloma. Although the survival of patients with IgD myeloma is shorter compared to other subtypes, the outcome of IgD myeloma is improving with the use of novel agents and autologous transplantation.35,36 The median survival of patients with IgD myeloma who received traditional chemotherapy was <2 years.9 Novel agents have improved the survival in these patients.5,17 Studies comparing bortezomib-based regimens with non-bortezomib-based regimens showed higher PFS and OS with bortezomib-based treatments.17
Blade et al.7 reported a median OS of 21 months, with 3-year and 5-year survival of 26% and 21%, respectively. In the AMN study, the median OS was 36.5 months. Patients who received immunomodulatory drugs had better OS, and patients who underwent autologous stem cell transplant (ASCT) had a median OS of 45.7 months.34 Kim et al.37 reported on 75 patients with IgD myeloma from the Korean Myeloma Registry. In the current study, all patients received bortezomib-based treatment, 50% of patients were alive at 2 years, and the median OS was 21 months. Table 3 gives a summary of clinical characteristics and outcomes of salient studies in IgD myeloma published in literature, and compared with the present study.
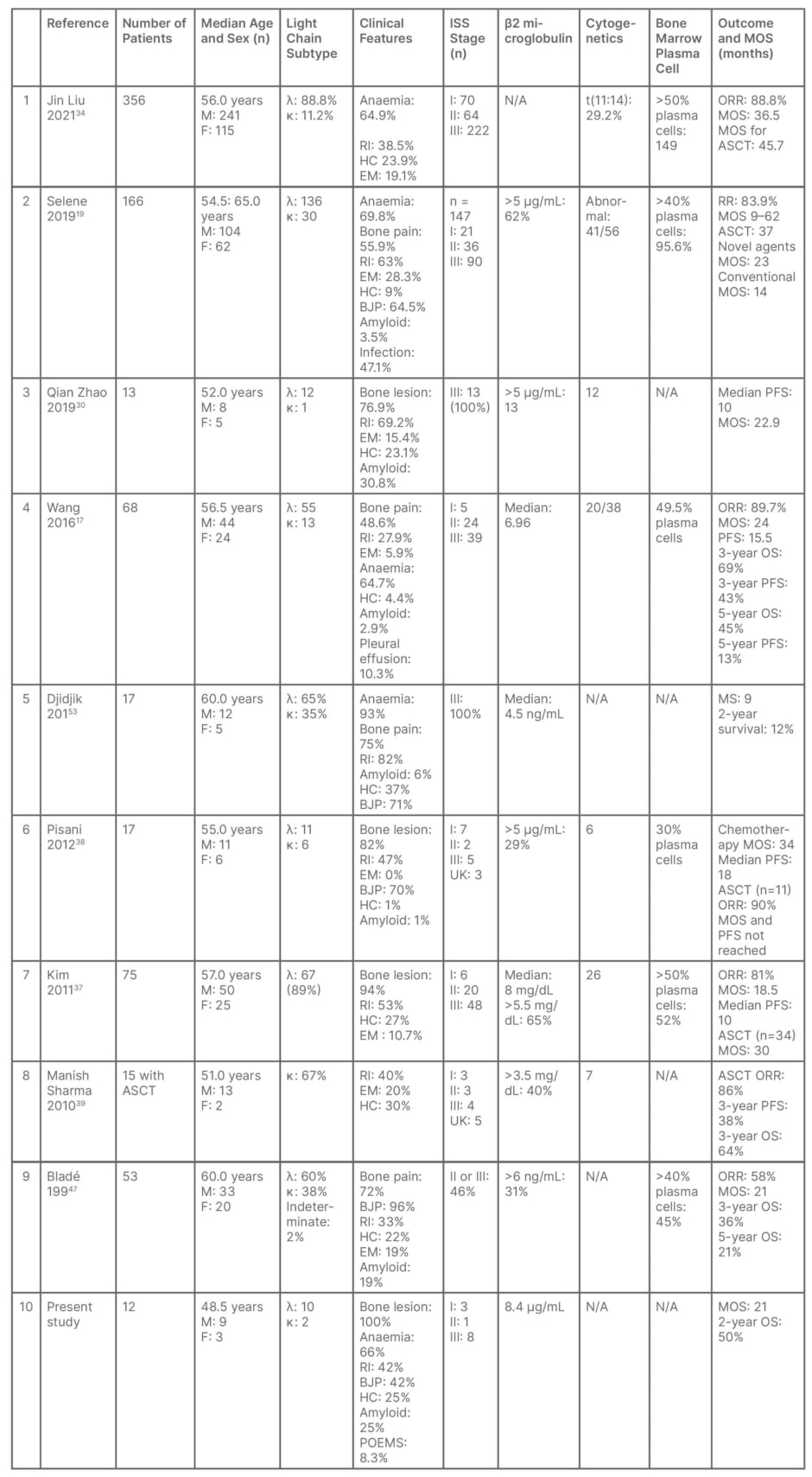
Table 3: Summary of clinical characteristics and outcomes of studies in immunoglobin D myeloma, published in literature.
ASCT: autologous stem cell transplantation; BJP: Bence–Jones protein; EM: extramedullary; F: female; HC: hypercalcaemia; ISS: International Staging System; M: male; MOS: median overall survival; N/A: not applicable; ORR: overall response rate; OS: overall survival; PFS: progression-free survival; POEMS: polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes; RI: renal insufficiency.
New generation agents pomalidomide, proteasome inhibitors carfilzomib and ixazomib, histone deacetylase inhibitors panobinostat and vorinostat, and monoclonal antibodies daratumumab and elotuzumab, may be promising in the treatment of IgD myeloma, but have not yet been verified in clinical trials. Studies comparing the outcomes following chemotherapy versus ASCT showed significant survival benefit in patients treated with ASCT.12,14,15 The use of high-dose chemotherapy followed by ASCT increased OS and PFS by 63% and 69%, respectively, compared to patients treated with conventional chemotherapy.38 However, a comparison of the outcome of patients with IgD myeloma who underwent ASCT with patients with non-IgD myeloma did not show any significant difference in PFS and OS.39 Venetoclax, a Bcl-2 inhibitor, might provide a new option for patients with t(11:14) translocation in view of the increased expression of BCL-2 in patients with IgD myeloma.34 Recently, sensitive quantitative IgD assays were shown to increase the PFS prediction accuracy in IgD myeloma.40 The relevant clinical role of laboratory in the detection of this rare disease was also highlighted by Intra et al.41
This study has some limitations. The authors do not have data on non-IgD myeloma from their institute; hence they are unable to compare the results of IgD and non-IgD myeloma. They are also unable to risk stratify based on cytogenetics, since the data were not available.
CONCLUSION
IgD myeloma is a rare subtype of MM, associated with a high incidence of renal failure, hypercalcaemia, extraosseous disease, amyloidosis, and Bence–Jones proteinuria. IgD λ disease is more frequent, unlike other subtypes of MM. Since IgD levels are generally very low in the serum, it may escape detection, and may lead to misdiagnosis as non-secretory myeloma. Immunofixation electrophoresis is very helpful in making the diagnosis, as in the authors’ case. Treatment is similar to other myeloma subtypes. Bortezomib-based chemotherapy is an effective treatment , with 50% of patients alive at 2 years. The outcome of IgD myeloma has improved, with the use of newer drugs, and with ASCT. More studies with a larger number of patients are needed to help understand the biology, and improve the outcome, of this rare myeloma subtype.