INTRODUCTION
Epidermodysplasia verruciformis (EV) is a rare, autosomal recessive genodermatosis affecting the skin. It is characterised by an unusual susceptibility to the human papillomavirus (HPV) infection, especially with HPV5 and HPV8, without gender and race preponderance.1,2 It can also be acquired and is more common in patients who are immunocompromised, such as those who have had a kidney transplant.3
The clinical feature is characterised by hypo- or hyperpigmented macules, skin-coloured flat-topped papules, and plaques with mild scaling that develop in children over the face, neck, trunk, and extremities.1,4 According to reports, the risk of malignant transformation of skin lesions, particularly those on sun-exposed areas, is increased by 35–50%.1,5
The diagnosis of EV is usually clinical, and it can be confirmed by specific histopathological findings and EV-HPV identification using PCR.6 There is currently no effective treatment for EV. However, early diagnosis, sun protection, life-long monitoring for malignant transformation, and therapeutic modalities such as acitretin, imiquimod, topical retinoids, cryotherapy, and others are available for the treatment of EV.7
Only a few cases of acquired EV, especially among recipients of a renal transplant, have been reported in the literature worldwide. The authors report a case of acquired EV in a patient who had a renal transplant, along with a review of the literature.
CASE PRESENTATION
A 24-year-old female patient who had had a renal transplant presented to the dermatology clinic with asymptomatic hypopigmented macules with branny scaling and multiple flat-topped shiny papules over the forehead and cheek bilaterally. She was on systemic corticosteroid and mycophenolate mofetil and had consanguineous parents.
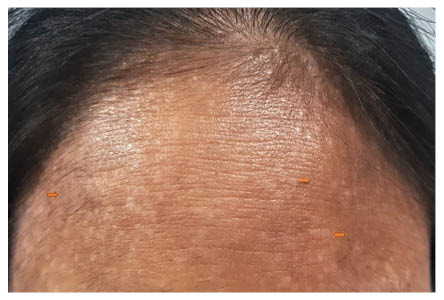
Figure 1: Multiple hypopigmented flat-topped papules and plaques present over the forehead (orange arrows).
The lesions started on the forehead, gradually increasing in number to involve the cheek. There were no similar skin lesions among family members. Photosensitivity, joint pain, skin rashes, oral ulcer, fever, night sweats, and lymphadenopathy were not present. On examination, there were numerous hypopigmented macules, with mild scaling as well as numerous skin-coloured flat-topped papules and plaques over the forehead and cheeks on both sides. The size ranged from 0.5 to 1.0 cm, the shape was round to oval, and the surface was smooth with normal surrounding areas (Figures 1 and 2). Hair, nail, and mucous membrane were not involved, and systemic examination was normal. The patient’s serological examination ruled out HIV infection.
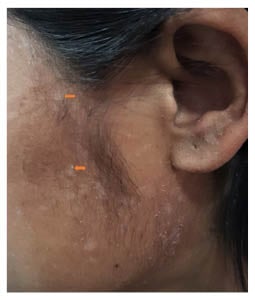
Figure 2: Multiple hypopigmented macules with scaling present over the lateral side of the face (orange arrows).
A punch skin biopsy was performed on the hypopigmented, slightly scaly macules over the forehead, which showed basket-weave hyperkeratosis; acanthosis; mild papillomatosis; prominent granular layer with vacuolisation of keratinocytes in the upper and lower epidermis; and a focal area showing enlarged or swollen keratinocytes in the upper layer, with pale bluish cytoplasm. The epidermal cells showed no dysplastic changes (Figure 3). Based on the clinical and histopathological features, a diagnosis of EV was made. The patient was given topical tretinoin gel and told to wear sunscreen on a daily basis, with regular follow-up.
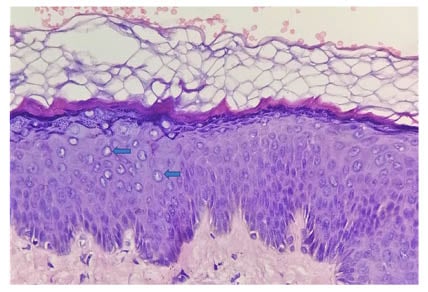
Figure 3: A histologic analysis of the forehead with a punch biopsy showed basket-weave hyperkeratosis, hypergranulosis, acanthosis, mild papillomatosis, and a large focal area affected with keratinocytes, with characteristic steel blue–grey cytoplasm, large nuclei, and clumping of keratohyaline granules (blue arrows) in the upper epidermis (haematoxylin-eosin stain; original magnification: ×40).
DISCUSSION
EV is a rare, autosomal recessive genodermatosis, which affects cell-mediated immunity and has no preference for gender, race, or geographic area.1 There is an increased risk of non-melanoma skin cancer, mainly squamous cell carcinoma (SCC).3 The disease can be sporadic, hereditary, or acquired, with the hereditary form being the most common. The acquired form can be seen in recipients of a renal transplant, Hodgkin’s disease, systemic lupus erythematosus, and HIV infection.3
On chromosome 17q25, the EVER1 or EVER2 genes have been linked to impaired cell-mediated immunity and an unusually high susceptibility to a specific strain of HPV infection.1 A higher lifetime cumulative sunlight exposure, X-ray irradiation, and immunologic defects in patients with EV are likely to induce mutations of the tumour suppressor gene protein (p53), leading to the development of skin malignancies in adult patients.8,9 Transformation into non-melanoma skin cancer occurs in 35–50% of patients between the ages of 40 and 50.4 Malignant transformation is caused by infection with specific strains of HPV, particularly HPV5 and HPV8. More than 30 EV-associated HPVs have been identified, including HPV5, HPV8, HPV12, HPV14, HPV15, HPV17, HPV19, HPV25, HPV36, HPV38, HPV47, and HPV50.3,4,10 Skin malignancies normally develop after a long period of time, and they seldom metastasise or penetrate deeper tissues.6
In patients with EV, hypo- or hyperpigmented macules resembling pityriasis versicolor, verruca plana-like lesions, and seborrhoeic keratosis-like plaques are prevalent. These lesions typically start in childhood and affect the face, neck, extremities, and trunk, with mucous membrane involvement being rare.1,3,9
The clinical and histopathological examinations are used to diagnose EV. All clinical lesions of EV share common histopathological features, which include basket-weave hyperkeratosis of the stratum corneum, parakeratosis, acanthosis, and characteristic cytopathic changes of infected keratinocytes in the malpighian layer. Infected keratinocytes show characteristic cytopathic changes characterised by large cells with pale blue–grey cytoplasm and variable sized kerato-hyaline granules. Regardless of the infecting HPV strain, these findings are consistent across all EV-HPV infections.6
There is currently no effective treatment for EV. There are, however, certain medical and surgical treatments available. Topical and oral retinoids, imiquimod, cimetidine, interferon-α, electrocautery, and cryotherapy are some of the options. All of these therapeutic options, however, are ineffective against EV. In order to prevent skin cancer, patients must be educated about the disease’s relapsing and persistent course, photoprotection, early detection, and excision of premalignant and malignant lesions with regular follow-up.3,8
CONCLUSION
Acquired EV is a rare entity and can be acquired in patients who are immunodeficient, especially in those who have had a transplant. Recipients of a renal transplant with acquired EV are at high risk of developing non-melanoma skin cancers.11 As a result, it is critical for the treating physician to recognise and accurately diagnose acquired EV as early as possible, and to reduce the risk of early malignant transformation of skin lesions through strict sun protection and regular follow-up.