
Abstract
Clinical laboratories worldwide are implementing next-generation sequencing (NGS) to identify cancer genomic variants and ultimately improve patient outcomes. The ability to massively sequence the entire genome or exome of tumour cells has been critical to elucidating many complex biological questions. However, the depth of information obtained by these methods is strenuous to process in the clinical setting, making them currently unfeasible for broader adoption. Instead, targeted sequencing, usually on a selection of clinically relevant genes, represents the predominant approach that best balances accurate identification of genomic variants with high sensitivity and a good cost-effectiveness ratio. The information obtained from targeted sequencing can support diagnostic classification, guide therapeutic decisions, and provide prognostic insights. The use of targeted gene panels expedites sample processing, including data analysis, results interpretation, and medical reports generation, directly affecting patient management. The key decision factors for selecting sequencing methods and panel size in routine testing should include diagnostic yield and clinical utility, sample availability, and processing turnaround time.
Profiling by default all patients with late-stage cancer with large panels is not affordable for most healthcare systems and does not provide substantial clinical benefit at present. Balancing between understanding cancer biology, including patients in clinical trials, maximising testing, and ensuring a sustainable financial burden for society requires thorough consideration. This review provides an overview of the advantages and drawbacks of different sizes NGS panels for tumour molecular profiling and their clinical applicability.
NEXT-GENERATION SEQUENCING
NGS, also known as massively parallel sequencing, has revolutionised cancer research and treatment, providing sensitive and accurate high-throughput platforms for large-scale genomic testing.1 The outstanding development of sequencing technologies has led to a rapidly growing body of pioneering research exploring the genomic landscape and molecular mechanisms of various cancer types, as well as the discovery of numerous genetic drivers of neoplastic growth (i.e., mutations that confer a selective growth advantage, thereby promoting cancer development). These genomic aberrations are single nucleotide variants, small insertions and deletions, copy number variations, and large gene structural variants, which accumulate in the genome during tumour development. While some mutations are already present at the time of diagnosis, others arise through clonal evolution during disease progression and are directly involved, for example, in mechanisms of treatment resistance.2 The abundance of genomic and transcriptomic sequencing data generated allows us to address a variety of previously impossible questions and offers numerous potential applications in precision medicine and drug discovery. Examples include transcriptomic studies that have been used to identify predictive disease-related expression patterns or expression differences due to drug side effects,3-5 as well as extensive genomic databases with curated information on DNA variants with clinical implications (e.g., OncoKB), which are often used as references for annotating observed variants.
In recent years, an increasing number of clinical laboratories worldwide are implementing NGS to detect genomic variants affecting the cancer genome. The main goal is to improve patient stratification strategies while supporting subsequent treatment decisions.
NGS strategies to identify genomic variations at the DNA level can be broadly divided into three main types: whole genome sequencing (WGS), whole exome sequencing (WES), and targeted sequencing (Figure 1). Sequencing cells’ whole genome or exome has been critical for answering complex biological questions. However, these methods provide a depth of information that is difficult to analyse and process in a clinical setting,6 making them currently unsuitable for broader application. The growing knowledge of the molecular alterations that initiate and drive tumour growth and metastasis has led to the development and the introduction of a variety of targeted gene panels that are used in routine clinical settings. Targeted sequencing, which typically involves analysis of a selection of genes with clinical significance, is by far the most widely used approach that best balances accurate identification of genomic variants with high sensitivity and a good cost-effectiveness ratio. Depending on the number of genes covered by the assay used, the targeted approach can be further subdivided into entry-level sequencing, characterised by the use of small- to medium-sized panels (i.e., up to 50 genes or similar) or comprehensive profiling based on large panels (in the range of hundreds of genes).6 Information obtained from targeted gene panels can inform diagnostic classification, guide therapeutic decisions, and/or provide prognostic insights for a given tumour.7 In addition, the use of targeted gene panels accelerates overall sample processing,1 including data analysis, interpretation of results, and generation of medical reports, which directly affects downstream patient management.
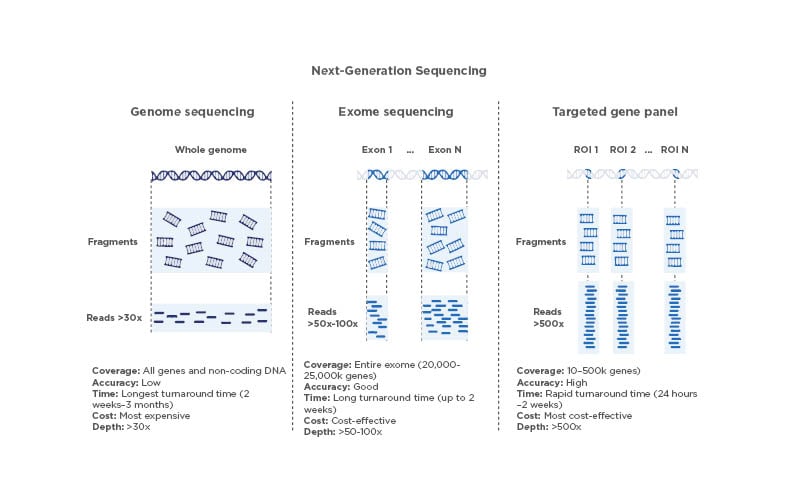
Figure 1: Comparison of next-generation sequencing techniques.
Different features of WGS, WES, and sequencing using targeted gene panels. The depth values shown are consistent with the established general recommendations for the sequencing approaches summarised here.
Image created with BioRender.com.
ROI: regions of interest; WES: whole exome sequencing; WGS: whole genome sequencing.
Targeted NGS can be performed using a variety of molecular methods to select for genomic regions of interest. The most common are hybridisation capture and PCR amplicon enrichment. The technologies behind these methods are different and bring their own advantages and challenges (Figure 2). The hybridisation capture-based approach is most commonly used for wide screening of genomic variants, often involving WGS and WES, and combined with high throughput sequencing. In contrast, the PCR amplicon-based approach has a simpler and faster workflow and is mainly suitable for targeted sequencing. The PCR amplicon-based method is of great utility for low sample volume input and is, therefore, commonly used in routine clinical testing.4
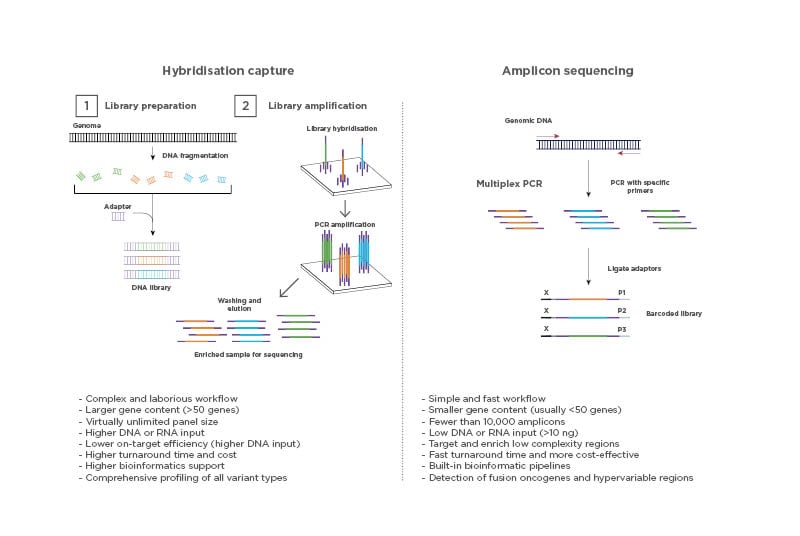
Figure 2: Types of next-generation sequencing-based assays.
Target enrichment for NGS can be performed using a hybridisation capture-based method or a PCR amplicon-based method.
Hybridisation capture can be performed either in a solution or on a solid substrate such as a microarray. Both methods require the use of synthetic oligonucleotide probes complementary to the genetic sequence of interest. The target regions are then amplified and washed to obtain the desired isolated regions for sequencing.
Amplicon-based enrichment uses carefully designed PCR amplicons to flank the target regions and specifically amplify the regions of interest. The amplified products are then purified from the sample and used for sequencing, eliminating the need for enrichment by hybridisation. Image created with BioRender.com
This review provides an overview of the advantages and drawbacks of NGS panels of different sizes for tumour molecular profiling and their clinical applicability. Molecular profiling using NGS, including current guideline recommendations is discussed here in the context of lung cancer, a pivotal example of precision medicine for disease diagnosis and treatment.
TUMOUR MOLECULAR PROFILING USING TARGETED NEXT-GENERATION SEQUENCING PANELS
Currently, molecular profiling of tumour samples plays a crucial role in the clinical management of patients with cancer. Rapid identification of genomic aberrations not only enables better stratification of patients for subsequent treatment with effective targeted therapies, but may also enable efficient disease monitoring and more accurate prognosis in the near future. Targeted NGS panels have been instrumental in this paradigm shift.2 When designing an NGS panel, it is important to understand the intended use. For example, if the goal is to screen for therapeutic targets and also to enrol patients in clinical trials for investigational therapies, large panels that allow testing for complex biomarkers such as tumour mutation burden or genomic instability (e.g., homologous recombination deficiency), are usually the preferred option. Depending on whether a test is designed for initial disease screening, which requires high sensitivity and high coverage, or for disease monitoring, which focus on specific mutations, the panel size used may also vary.8 Overall, the selection of the number and type of genes in a given NGS panel requires careful consideration. The size of the panel (number of genes and extent of gene coverage); type of samples to be tested; turnaround time and sensitivity required; type and complexity of variants to be determined; extent of bioinformatics support, including infrastructure and laboratory resources; and available technical expertise must be considered by the testing laboratory before selecting an NGS solution for routine clinical service.7 Some of these parameters and the advantages and disadvantages of choosing different panel sizes (Table 1) are discussed in the following sections.
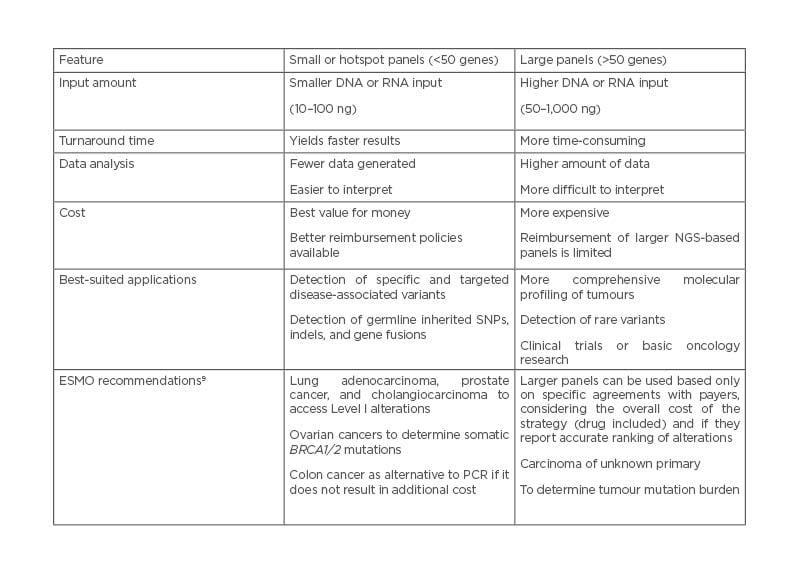
Table 1: Comparison between small or hotspot, and large next-generation sequencing-based gene panels.
ESMO: European Society for Medical Oncology; NGS: next-generation sequencing; SNP: single nucleotide polymorphism.
Sample Quality and Quantity Requirements
Regardless of the NGS approach and methodology to be used, the feasibility of molecular profiling depends on the quality and quantity of the sample to be tested. The use of NGS to detect low allele frequency somatic variants in nucleic acids extracted from formalin-fixed paraffin-embedded tumour tissue is challenging for clinical molecular diagnostic laboratories, because these types of samples often yield low quantities of degraded, poor-quality genetic material.10,11 It is estimated that molecular profiling fails in 5–30% of tested patients due to insufficient material or poor sample quality,12,13 with the hybridisation capture method being the most affected by this issue. Thus, correct preservation and handling of clinical samples are critical for the application of panel testing in daily practice. The quality of nucleic acids extracted from formalin-fixed paraffin-embedded samples is negatively affected by several factors, including tissue age, fixation time, and tumour lesion size (i.e., large tumour masses often have hypoxic areas leading to high cell death).11 In addition, the quantity of nucleic acids may be limited by the tumour sampling methods (e.g., fine-needle aspiration) and the tumour cellularity.14,15
Another aspect to consider is that most patients with cancer are only diagnosed at advanced stages, when the available sample material is often limited.16 This situation per se hampers the use of large gene panels, an approach that may require a large amount of sample material to provide fully reliable results. The minimum sample input requirements for NGS depend on the enrichment technique, sequencing method, and depth of sequencing required. In particular, when using small- to medium-sized targeted gene panels, successful profiling is possible with as little as 5–10% of neoplastic cell sample content and 10 ng of DNA or RNA.17 This is particularly relevant when the amount of biopsied tissue is reduced, as in the case of fine-needle aspirates.
The desired limit of detection must also be considered to determine the minimum amount of DNA or RNA required to perform a test and the lowest frequency of mutant alleles that can be detected.18 This is important for all oncology assays, where tumour percentage and heterogeneity affect mutation allele frequencies.19
Bioinformatics and Interpretation of Variants
The large amount of raw data generated by NGS-based assays requires a bioinformatics pipeline capable of converting nucleotide sequences into meaningful biological and clinically actionable results. In addition, such an analysis must meet several analytical requirements and ensure the accuracy and reproducibility of the results obtained.
A typical pipeline for analysing NGS data can be divided into four main operations: base calling, read alignment, variant identification, and variant annotation.20 The larger the region of the sequenced genome, the greater the likelihood of encountering rare or novel variants that require complex interpretation. As the size of diagnostic panels increases, the likelihood of detecting incidental findings also increases.19 The available interpretation software is extensive and is constantly updated as new biomarkers are discovered and NGS assays are introduced to the market.18,21,22
Another challenge is deciding which genes to test in a given clinical scenario. Although there are guidelines that define the most common mutations or genes of interest (tests that are usually reimbursed), the literature and clinician interest may propose other genes (tests that are usually not reimbursed) that may be medically useful.19,23 To standardise the reporting and interpretation of clinically relevant genomic data in the management of patients with cancer, the European Society for Medical Oncology (ESMO), led by the ESMO Translational Research and Precision Medicine Working Group, developed the Scale for Clinical Actionability of molecular Targets (ESCAT) ranking system.24 ESCAT defines six levels of evidence as Tier I: targets ready for implementation in routine clinical decisions; Tier II: investigational targets that likely define a patient population that benefits from a targeted drug but magnitude of benefit is unknown; Tier III: clinical benefit previously demonstrated in other tumour types or for similar molecular targets; Tier IV: preclinical evidence for actionability; Tier V: evidence supporting co-targeting approaches but without clinical benefit; and Tier X: lack of evidence for actionability. In addition, organisations such as the Association for Molecular Pathology (AMP), American Society of Clinical Oncology (ASCO), and College of American Pathologists (CAP) have also developed classification systems to rank the clinical utility of variants based on the level of evidence.
In particular, for WGS and WES, performing the entire workflow from DNA or RNA extraction to bioinformatics analysis, requires trained personnel with extensive expertise and considerable hands-on laboratory work. On the other hand, several targeted solutions with fully automated end-to-end workflows are commercially available. They offer optimised solutions for data analysis and reporting that can be easily handled by a certified technician with a minimum of NGS-specific training.
Diagnostic Yield and Clinical Utility
The diagnostic yield is an important selection criterion for determining the performance of any assay, including those for genetic testing. It is primarily defined as the likelihood that a test will provide the required information for a genetic diagnosis. For example, laboratories can use this parameter to decide whether to switch from one particular test method to another (e.g., fluorescence in situ hybridisation for gene fusion detection versus RNA-based NGS). The same is true when deciding between sequential testing for single genes and NGS, as in the example reported by Pósafalvi et al.,25 showing that using a targeted panel of 55 cardiomyopathy genes significantly improved the percentage of patients in whom disease-causing mutations were found (15–50% in more than 250 patients tested), demonstrating the benefits of adopting NGS-based methods in clinical diagnostics.
In another study, Garg et al.26 compared the clinical diagnostic yield of a targeted sequencing panel (Trusight Tumor 26 [Illumina, San Diego, California, USA]) in melanoma, colorectal, and gastrointestinal stromal tumours with non-NGS assays. Overall, 79% of melanomas and 94% of colorectal tumours were positive on panel testing, demonstrating that NGS increases diagnostic yield by 24% and 36%, respectively, for routinely tested variants in melanoma and colorectal cancer. No additional benefit was observed in gastrointestinal stromal tumours. Nevertheless, these results demonstrate the impact of NGS-based assays on diagnostic yield in cancer.26
In a study aimed at understanding the appropriate size of a solid tumour sequencing panel to identify clinically actionable variants, Vail et al.27 directly compared the results of a large gene panel (315 genes, reference laboratory assay [Foundation Medicine, Cambridge, Massachusetts, USA]) with those of a medium-sized panel (161 genes, Oncomine™ Comprehensive Assay [Thermo Fisher Scientific, Waltham, Massachusetts, USA]) and a small hotspot panel of 50 genes (Oncomine™ Precision Assay [Thermo Fisher Scientific, Waltham, Massachusetts, USA]).27 While the larger panel detected more variants, the additional variants beyond those included in the medium panel had no impact on patient management. The data indicate that any variant identified by the larger panel in the context of an U.S. Food and Drug Administration (FDA)-approved therapy would also have been identified by the medium panel. Even more remarkably, nearly all variants (88.5%) would have been identified by the 50-gene panel. Overall, this comparative analysis of the clinical utility of gene panels of different sizes shows that small and medium-sized optimised gene panels are as informative as larger panels when the primary goal is to identify clinically actionable mutations.27
The use of diagnostic yield as a criterion also implies that genes included in a panel should be carefully selected (i.e., only genes for which sufficient data are available to demonstrate their involvement in the disease of interest).28 Considering the ESCAT ranking, prevalence of alterations, number of patients to be tested with NGS, and matching of an effective drug in routine clinical practice, ESMO reported that there was no proven public health impact if actionable alterations were detected beyond ESCAT Level I.24 In light of this, ESMO recommends larger panels to be used only based on specific agreements with payers, taking into account the total cost of the patient treatment strategy (including drugs) and when they provide an accurate ranking of mutations.28 Overall, ESMO suggests that from a public health perspective, targeted small- to medium-sized NGS panels should be the primary choice in patients with the following metastatic cancers: advanced lung adenocarcinoma, prostate cancer, ovarian cancer, and cholangiocarcinoma (Table 1).9,29 Patients with other cancers could decide with their physician to order NGS for a large panel of genes, provided that there is no additional cost to the public healthcare system and the patient is informed of the relative likelihood of benefit (patient-centric perspective). Similarly, large gene panels should be used in academic reference centres where clinical and translational research is conducted.9,29
Turnaround Time and Cost-Effectiveness
One of the most critical components of clinical testing where rapid decisions must be made is the turnaround time of the test. Oncologists need to start treating patients quickly, especially if the cancer is aggressive or refractory.30 Larger gene panels are more time consuming as they may require a more complex data analysis workflow. In contrast, small hotspot panels (<50 genes) or medium-sized panels are best suited to achieve faster results because they are less ‘sequencing intensive’ and their analysis is based on a limited number of clinically valuable targets.16 Any solution that can deliver fast and reliable results should ideally be considered for routine testing. The consequences of a long testing turnaround time can have a negative impact on the clinical management of patients with advanced-stage cancer, with reduced outcomes, and should be avoided (2022 ASCO Annual Meeting abstract, unpublished data in press).
Another important component of diagnostic testing is cost, which is directly influenced by several components, such as the choice of enrichment strategy used (hybridisation capture versus PCR amplicon), size of the genome targeted, and labour and equipment required for data generation and analysis.19 The cost of library preparation and overall sequencing also depends on samples’ batching. With current NGS platforms, the data yield is high enough to barcode multiple samples and sequence them together (i.e., multiplexed). However, the size of a targeted gene panel (i.e., the number or size of genes targeted) determines the degree of multiplexing that can still achieve good coverage per sample (i.e., large panels have a low degree of multiplexing and small panels have a high degree).30 Sequencing solutions are now available that allow cost compression to combine small- to medium-sized targeted panels with optimised sample batching capabilities.31
THE PARAMOUNT EXAMPLE OF NEXT-GENERATION SEQUENCING TESTING UTILITY: LUNG CANCER
Lung cancer remains one of the most commonly diagnosed cancers and the leading cause of cancer mortality worldwide.32 Among histologic types, non-small cell lung cancer (NSCLC) is the most common and accounts for approximately 80–85% of all cases, of which about 40% are adenocarcinoma, 25–30% are squamous cell carcinoma, and 10–15% are large cell carcinoma.33 Recent advances in characterising the molecular biology of lung cancer, particularly the discovery of genetic driver mutations in non-squamous NSCLC, have led to the development of unique targeted therapies that often achieve remarkable success in patients with these genetic alterations.29
Current guidelines published by AMP, CAP, and the International Association for the Study of Lung Cancer (IASLC) recommend testing for mutations in four to eight specific genes for each patient with NSCLC.27,34 This may result in the need for multiple tests using different techniques (e.g., fluorescence in situ hybridisation, immunohistochemistry, and Sanger sequencing), each with different tissue requirements and demanding different expertise.27 In this context, NGS panels have gradually replaced these techniques in clinical laboratories and allowed simultaneous analysis of multiple genes.20 The use of hotspot testing in advanced NSCLC, particularly through PCR amplicon-based methods that focus on unique gene alterations such as single nucleotide variants, indels, or gene fusions associated with effective targeted therapy, has been shown to be a cost-effective approach that can achieve high success rates (above 90%) even with low input material.35-37 In addition, extremely fast turnaround times (within 3 business days) have been reported, even for tests performed in a community hospital.38 Although the utility of large-panel NGS sequencing as an initial testing approach for all patients with NSCLC remains to be demonstrated,39 it is important to consider this testing option for patients who are driver-gene negative, as this may increase their chances of enrolment in clinical trials.
Clinical Practice Guidelines for the Use of Next-Generation Sequencing
The National Comprehensive Cancer Network (NCCN) Clinical Practice40 and ESMO guidelines41 for advanced NSCLC recommend molecular testing for clinically relevant biomarkers such as EGFR, BRAF, and KRAS mutations; ROS1, ALK, and NTRK1/2/3 fusions; MET exon 14 skipping mutations; and programmed death-ligand 1 expression. Emerging biomarkers include MET amplification, RET fusions, and ERBB2 (HER2) mutations.41
ESMO has proposed three levels of recommendations for the use of NGS in advanced non-squamous NSCLC based on the ESCAT ranking. It is recommended that a tumour (or plasma) sample from a patient with advanced non-squamous NSCLC could be profiled using NGS technology to detect ESCAT Level I alterations (i.e., targets suitable for implementation in routine clinical decisions such as EGFR, MET, BRAFV600E, ROS1, and ALK). In addition, the ESMO guidelines state that there is no evidence that panels detecting genes with a lower level of evidence add value from a public health perspective and should only be considered from a translational research perspective.9
CONCLUSIONS
NGS has become a robust, reproducible, and cost-effective technology to screen tumours for genetic alterations. The democratisation of NGS (i.e., its broad application from large academic centres to community hospitals) is critical to fully unleash the promise of ‘precision medicine’. Nevertheless, there are many approaches to perform NGS testing today, encompassing large to small sequencing assays, and several aspects must be considered when selecting a gene panel. Choosing the best panel size for clinical practice has sparked intense debate among researchers and clinicians. For routine patient testing, diagnostic yield and clinical utility, along with technical aspects such as sample availability and turnaround time, should be the main guiding principles to make the most appropriate sequencing method and panel size decisions. A cost-effectiveness analysis should be carefully considered, as testing per default all patients with late-stage cancer with large panels is not affordable for most healthcare systems worldwide, nor does it currently provide substantial clinical benefit for all patients. The need to advance our understanding of cancer biology and provide patients with the opportunity to participate in clinical trials, while ensuring that the financial burden remains reasonable, requires a thorough consideration of what panel size will best serve the target population of patients. Equally, considering the evolution of the ‘hub and spoke’ model in the context of current trends in social medicine, it is reasonable to think that the backbone of decentralised testing in regional and community hospitals could be represented by an NGS panel of smaller size, whereas testing in major reference hubs should include broader use of comprehensive genomic profiling.
To date, as there is no ‘one size fits all’ solution for either small- to medium-sized or large NGS panels, flexibility remains key. Nevertheless, the improvement in cancer knowledge achieved by wide mutation panels performed on a large scale could help prioritise some alterations for the design of ‘pragmatic’ and cost-effective panels for daily oncology practice in the future. Ultimately, what really matters is that as many patients with cancer as possible are tested effectively and reliably.






