Abstract
Diabetic nephropathy (DN) is a lethal microvascular complication associated with Type 1 and Type 2 diabetes mellitus, and is the leading single cause of end-stage renal disease. Although genetic influences are important, epigenetic mechanisms have been implicated in several aspects of the disease. The current therapeutic methods to treat DN are limited to slowing disease progression without repair and regeneration of the damaged nephrons. Replacing dying or diseased kidney cells with new nephrons is an attractive strategy. This review considers the genetic and epigenetic control of nephrogenesis, together with the epigenetic mechanisms that accompany kidney development and recent advances in induced reprogramming and kidney cell regeneration in the context of DN.
INTRODUCTION
Diabetic nephropathy (DN) is one of the major microvascular complications associated with diabetes in terms of increased healthcare costs, high morbidity, and premature mortality.1 More than 50% of patients with Type 2 diabetes mellitus (T2DM) and 30% of those with Type 1 diabetes mellitus (T1DM) develop kidney disease, and a considerable number of cases can progress to end-stage renal disease (ESRD).
In addition, diabetic patients with ESRD are more likely to develop adverse macrovascular complications such as hypertension, atherosclerosis, and peripheral and cerebrovascular disease leading to an increased mortality rate. DN is clinically characterised by progressive albuminuria, decreased glomerular filtration rates (GFR), and a constant decreased kidney function. It is histologically defined by renal glomerular hypertrophy, expansion of mesangial and tubular compartments, accumulation of mesangial extracellular matrix proteins, and podocytopenia associated with foot process effacement.2
DN is a complex multifactorial disease caused by multiple genetic and environmental factors. Genetics alone cannot fully explain the variability in the incidence of nephropathy and the uneven distribution and graveness of complications in diabetic patients.3 More recent studies have shown that epigenetic mechanisms are involved in the pathogenesis of DN. These processes influence gene expression patterns or cellular phenotypes and disease states with no underlying change in DNA sequence.4 Acute hyperglycaemia leads to chronic metabolic and haemodynamic derangements,5 which trigger chromatin structural changes, transcription factor activation, and gene expression.6 These changes persist even after returning to normoglycaemia:7 a phenomenon referred to as ‘metabolic memory’ or ‘legacy effect’.8 Thus, environmentally-induced epigenetic events compounded by genetic predisposition play significant roles in diabetes and its related complications (Figure 1).6
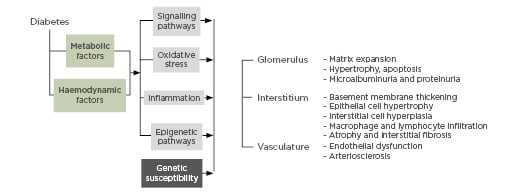
Figure 1: Pathobiology of diabetic nephropathy.
DKD: diabetic kidney disease
Identifying novel approaches for the prevention and treatment of DN relies on an improved understanding of the molecular mechanisms driving DN.9 The current review examines the emerging evidence for epigenetic mechanisms and pathways in DN. In addition, the authors also review future strategies in DN treatment such as transcriptional reprogramming of mature adult kidney cells into uncommitted induced pluripotent stem cells for renal repair and therapeutics. Key transcription factors involved in DN that can be targeted to halt disease progression are also described. A comprehensive understanding of the different molecular mechanisms driving DN is crucial to identify new therapeutic targets and potential biomarkers.9
PATHOBIOLOGY AND EPIGENETICS OF DIABETIC NEPHROPATHY
DN is a lethal microvascular complication associated with T1DM and T2DM and is the leading single cause of ESRD. DN is defined by a progressive increase in the urinary albumin excretion rate accompanied with an increased blood pressure and decline in GFR, with end-stage renal failure as the final endpoint. DN affects one-third of patients with T1DM and rarely develops before 10 years of diabetes duration, whereas in T2DM the prevalence ranges from 25% in patients younger than 65 years old to almost 50% in individuals older than 65 years.10 The first clinical sign of DN is an increased urinary albumin excretion rate in the range of ≥20 to <200 μg/min, or ≥30 to <300 mg/24 hours, termed microalbuminuria. Overt DN or macroalbuminuria is classified if the albumin excretion rate is ≥200 μg/min or ≥300 mg/24 hours, and is typically followed by a decline in kidney function, renal impairment, and ultimately ESRD (Table 1).
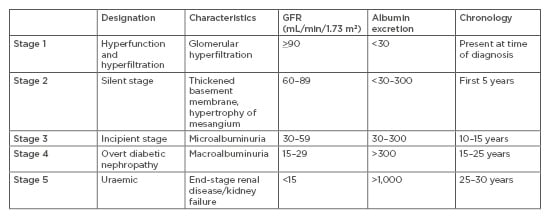
Table 1: Classification of diabetic nephropathy.
Table adapted from Haneda et al.11
GFR: glomerular filtration rate.
Chronic DN is the most common cause of renal replacement therapy in Western society.12
It is characterised by a decline in renal function, measured directly by serum creatinine, calculated creatinine clearance, or GFR. Renal function is classified based on GFR: normal renal function is considered as GFR ≥90, mild decrease in renal function is 60–89, moderate decrease in renal function is 30–59, severe decrease in renal function is 15–29, and renal failure <15 mL/min/1.73 m2.13
DN is associated with abnormalities in renal cell types including tubular and glomerular cells. Morphological changes occur in the glomeruli in individuals with DN. Among these changes, dysfunction of glomerular podocytes is critical for the subsequent development of glomerulosclerosis and nephron dropout.14
With the expectation of somatic mutation events, an individual’s DNA sequence is identical across different cell types; however, each has its own unique phenotype attributed to changes in gene expression. Epigenetics refers to the covalent modification of DNA and sequence-specific targeting of mRNA to control a cell’s phenotype via changes in gene expression. Epigenetic DNA modifications are heritable, reversible, and do not change the DNA sequence, but rather alter DNA structure and stability, replication, and transcription. The true genome-wide assessment of epigenetic modifications utilising next-generation technology is referred to as epigenomics.
Epigenetic Model for Human Disease or the Epigenetic State
The mechanism by which the epigenetic state of a normal cell is established can be defined and divided into three broad stages or mechanistic signals.15 The epigenetic state involves the interplay of an epigenator, which can be an environmental cue or trigger for the cell that precedes any modification to that cell’s epigenome. Next, an epigenetic initiator, which includes DNA-binding proteins and noncoding RNA, translates and coordinates specific responses conferred by the epigenator. The epigenetic initiator may not dissipate after its action, but rather persists with the maintainer. Lastly, the epigenetic maintainer, i.e., persistent marks such as DNA methylation and post-translational histone modifications, enable influence on structure and function of the genome, including when and where genes are transcriptionally activated or deactivated. Maintainers may function by carrying an epigenator signal through the cell cycle or could maintain epigenetic landscapes in terminally differentiated cell types.15
Aberrations at any stage of this system can potentially lead to subtle phenotype variations and molecular variations associated with a diverse range of human pathologies, including cancer, cardiovascular disease, and diabetes.16
The best characterised maintainer of the epigenetic state is DNA methylation. Considered a stable epigenetic mark, DNA methylation regulates gene expression and genome organisation and is transmissible from cell to cell, and in some instances is hereditary.17 In mammals, it involves the covalent addition of a methyl group to cytosine-phosphate-guanine (CpG) dinucleotides from the methyl donor S-adenosylmethionine and catalysis by DNA methyltransferases. The regions with higher CpG density, termed CpG islands, are often found around gene promoters and are usually methylated in a tissue-specific manner.18 Generally, a low methylation status of CpG islands at promoter sites is associated with gene activation, while transcription is repressed under a high DNA methylation status.19 Gene regulation is critically important for normal functioning of the genome. To this end, even genes that carry no mutations or disease-predisposing polymorphisms can be considered harmful if they are not expressed at the appropriate level in the correct type of cell at the right time. Aberrant DNA methylation patterns in key regulatory elements of the gene, such as promoters and enhancers, regulate gene expression.19 Classically, increased levels of gene methylation result in transcriptional repression21 and changes in DNA methylation have been found in human diseases, including allergies,22 cancer,23 T1DM,24 metabolic diseases,25 and cardiovascular diseases.26 Several models have been proposed for the molecular mechanisms of DNA methylation in gene regulation, in which one model proposes that aberrant DNA methylation at promoter regions can alter transcription of disease promoting and protective genes coordinated by methyl-CpG-binding domain proteins and transcription factor binding (Figure 2).
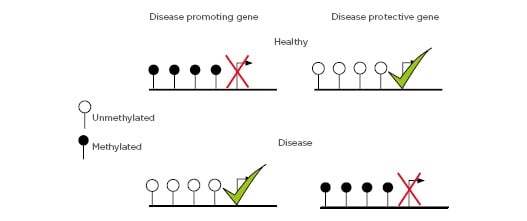
Figure 2: DNA methylation at disease promoting and protective genes.
Depiction of gene regulation mediated by DNA methylation at gene promoters.
DNA Methylation and Type 1 Diabetes Mellitus
In the last decade, experimental evidence has exemplified the importance of DNA methylation as a key mechanism by which the environment influences and interacts with genetic factors in the development of T1DM. For example, prospective analysis was performed on a cohort of nondiabetic monozygotic twins of patients with T1DM.27 In this analysis, a median discordance time of 4.2 years was observed in the 47 twins (25%) who had become concordant on follow-up.27 These results imply that genetic and nongenetic factors could contribute to the development of T1DM. Several studies in twin-pairs have measured changes in DNA methylation at CpG sites in immune effector cells, including monocytes and peripheral lymphocytes, as well as whole blood samples with an aim of identifying nongenetic and underlying epigenetic mechanisms influencing the development of T1DM. Rakyan et al.28 identified 132 differentially methylated CpG sites in T1DM-affected co-twins, of which 74 were hypomethylated and 58 were hypermethylated genes including GAD2 and HLA-DQB1, previously described in T1DM. Interestingly, T1DM-associated methylation was also detected in islet autoantibody genes GAD65 and IA-2. A similar study of monozygotic twins by Stefan et al.29 assessed changes in DNA methylation in DNA isolated from lymphoblast cell lines from three pairs of monozygotic twins discordant for T1DM and six pairs of monozygotic twins concordant for T1DM. They identified 88 CpG sites with significant changes in DNA methylation between all T1DM-discordant monozygotic twin pairs.29 Functional analysis suggested that differentially methylated genes were clustered in the immune response and defence response pathways.
A more recent twin study, using the Infinium® HumanMethylation450 BeadChip (Illumina, San Diego, California, USA), profiled changes in whole blood DNA methylation in twin pairs discordant for T1DM.30 The authors reported modest DNA methylation differences (range: 2.2–5.0%) for the major histocompatibility complex region and T1DM-associated CpG sites in BACH2, INS-IGF2, and CLEC16A. Other genes reported to have differential DNA methylation were MAGI2, FANCC, and PCDHB16. These findings are indicative of global DNA hypomethylation within gene promoter regions which may contribute to T1DM; however, the results do not show large DNA methylation differences at CpG sites between T1DM-affected and unaffected twins. More recently, a more comprehensive epigenome-wide association study across 450,000 CpG in 52 monozygotic twin pairs discordant for T1DM in three immune effector cell types showed substantial enrichment of differentially variable CpG positions (DVP) in T1DM twins when compared with their healthy co-twins and with healthy, unrelated individuals.31 A total of 10,548 DVP were identified in B cells, 4,314 in T cells, and 6,508 in monocytes. DNA methylation differences between the T1DM twin and their healthy co-twin were found to be comparatively large in many cases.31 Functional annotation and integration with cell type-specific gene regulatory circuits highlight pathways involved in immune cell metabolism and the cell cycle, including mTOR signalling. Interestingly, in the same study T1DM-associated DVP identified in T1DM-twins were measured in the cord blood of newborns who progressed to overt T1DM. This comparison revealed no statistically significant overlap between cord blood and T1DM-twins, but the results suggest that changes in DNA methylation likely emerge after birth.
DNA methylation patterns have also been suggested to be affected by hyperglycaemia. In a study using zebrafish as a model of hyperglycaemic memory, the authors demonstrated DNA hypomethylation was heritable.32 Hyperglycaemia was induced in adult zebrafish before entering a recovery phase. However, hyperglycaemia-induced global DNA hypomethylation was seen in the daughter cell tissue that did not have prior exposure to hyperglycaemia.
In patient populations, the large-scale prospective Diabetes Control and Complications Trial (DCCT)/Epidemiology of Diabetes Interventions and Complications (EDIC) and follow up studies33,34 have signified that the establishment of tight glycaemic control is critical for long-term control of T1DM diabetes. The findings also emphasise that poor glycaemic control, even when followed by intensive therapy, can mediate sub-inflammatory conditions and vascular complications. Recently, DNA methylation was studied at specific loci over two different time points in individuals from the DCCT/EDIC T1DM cohort.35 Chen et al.36 measured DNA methylation of approximately 450,000 CpG sites in genomic DNA of whole blood isolated at EDIC Study baseline from 32 cases (DCCT conventional therapy group subjects showing retinopathy or albuminuria progression by EDIC Study Year 10) versus 31 controls (DCCT intensive therapy group subjects without complication progression by EDIC Study Year 10). DNA methylation was also profiled in blood monocytes of the same patients obtained during EDIC Study Years 16–17. Comparing DNA methylation profiles of whole blood from the cases versus control, 53 regions showed hypomethylation and 225 hypermethylation, whereas in monocytes, 155 regions were hypomethylated and 247 hypermethylated. Notably, only 12 differentially methylated regions were common to both cell populations, including thioredoxin-interacting protein, known to be associated with hyperglycaemia and related complications.35
The authors also found a set of differentially methylated regions represented similar trends of associations with prior HbA1c in both whole blood monocytes. Follow-up experiments in monocytes showed that high glucose induced similar persistent region-specific hypomethylation at the thioredoxin-interacting protein region and this was inversely correlated with gene expression. This study provides evidence that hyperglycaemia can mediate changes in DNA methylation which persist for several years. In addition to the work described, the authors have also shown that hyperglycaemia regulates genome-wide DNA methylation signatures in primary vascular cells.36 These studies highlight the importance of glucose exposure in DNA methylation and that consideration should be made while analysing methylation data from patients with diabetes.
DNA Methylation and Diabetic Microvascular Complications
T1DM is associated with multiple macrovascular and microvascular complications leading to increased morbidity and mortality. While studies have shown that DNA methylation is associated with the pathogenesis of diabetes, there is emerging evidence that it also may contribute to the development of diabetic complications in peripheral organs such as the kidneys, retina, and peripheral nerves.
Increasing evidence suggests that changes in DNA methylation are involved in DN.37 Epigenetic mechanisms have been proposed by which protection occurs in some individuals with diabetes, or through which some individuals with diabetes seem predisposed to progressive chronic kidney disease. One clinical study showed that whole blood genomic DNA from T1DM patients with DN exhibited differential DNA methylation patterns at 19 genes including UNC13B, relative to those without nephropathy.38 A large case-control association study undertaken in T1DM individuals with or without DN employing the 450,000 and 27,000 methylation arrays identified 54 differentially methylated probes across 51 unique genes in blood-derived DNA. A sub-analysis, assessing DNA methylation in individuals with ESRD (versus without DN) revealed the detection of 755 differentially methylated probes in 374 genes. Of interest, 43 of the top-ranked genes for DN were also identified in the subgroup of patients with ESRD.39 Pathway analysis of top-ranked genes revealed an association with metabolic pathways and mitochondrial function implicated in DN. DNA methylation profiles in proximal tubules obtained from db/db mice uncovered differentially methylated gene targets implicated in glucose metabolism and transport, leading to a resistance to the effects of pioglitazone.40
CONTROL OF CELL IDENTITY IN KIDNEY DEVELOPMENT AND THERAPEUTICS
The adult mammalian kidney cannot sufficiently regenerate or replace damaged kidney tissue with new nephrons after injury.41 Given the drastic shortage of donor kidneys for transplantation, this calls for urgent development of novel regenerative therapies to reverse the damage caused by T1DM on the kidney.
The latest discoveries in the fields of developmental nephrology hold great promise for kidney regenerative medicine, enabling researchers to design novel therapeutic tools and approaches to regenerate nephrons for DN. To advance kidney therapeutics further, it is mandatory to gain a deeper understanding of the key cellular and molecular programmes involved in nephrogenesis and kidney regeneration. All cells in the body arise from embryonic precursors through the coordinated activity of trans-acting transcriptional regulators and cis-acting modifications in DNA. These transcription factors act at multiple stages of kidney development, and adult kidney function or repair. Many of these transitions governing cell identity involve changes in gene expression. This in turn is regulated by epigenetic processes, including DNA methylation and histone modifications.
However, the therapeutic targeting of DNA methylation and transcriptional control in DN are understudied; therefore, a focus on histone modification has become the preferred option.
HISTONE MODIFICATIONS AS A THERAPEUTIC MODALITY IN DIABETIC NEPHROPATHY
The human kidney has a complex internal organ system and is composed of highly diverse cell types, including epithelial, stromal, and endothelial cells.42 All of these must be assembled into discrete anatomic and functional structures at the earliest embryonic stages. The human kidney arises from an embryonic structure known as the metanephros, the last of the three excretory organs (pronephros, mesonephros, and metanephros) to develop from the intermediate mesoderm around embryonic Day 10.5, namely the metanephric mesenchyme (MM) and the ureteric bud (UB).43 On entering the metanephric mesenchyme, cell interactions between the UB and adjacent mesenchyme drive the assembly of the functional kidney. At the same time, the UB induces the condensation of MM cells to form cap mesenchyme. Cap MM cells contain the progenitors/stem cells of the nephrons identified by their expression of Six2.43 While the UB gives rise to renal collecting ducts, the condensed cap mesenchyme gives rise to a population of stem/progenitor cells that undergo mesenchymal–epithelial transition originating nephrons.43 At 34 weeks of gestation in humans, nephron progenitors cease propagation and are terminally differentiated with no cell renewal/replication capability, and thus no nephron formation occurs in the adult kidney, underlying the irreversible nature of DN.44 Histone modifications are closely linked to nephron differentiation. Nephron progenitors feature equally enriched active and repressive marks (H3K4me3, H3K9me3, and H3K27me).45
Over a decade ago, the seminal work by Takahashi and Yamanaka46 showed that ectopic expression of key transcription factors Oct4, Sox2, Klf4, and c-Myc (OSKM cocktail) could reprogram differentiated cells to pluripotency.46 Induced pluripotency is a process characterised by gradual changes in the epigenetic landscape.47 Understanding how the reprogramming factors alter the cell epigenome to reset cell identity represents an important aim for the regenerative medicine field. Successful reprogramming to induce pluripotency is largely dependent on faithful remodelling of the cell’s epigenetic states to silence gene expression and activate the transcriptional machinery characteristic of pluripotent cells. Trans-differentiation, also known as lineage reprogramming, is a process in which one somatic cell transforms into another mature somatic cell bypassing the pluripotent state. A number of mammalian programming strategies have recently been described, i.e., Al-Hasani et al.48 have demonstrated that the paired box (Pax) 4 protein as well as γ-aminobutyric acid convert adult α-cells (glucagon) in pancreatic islets into functional β-like insulin producing cells in vivo.49 Epigenetic modification with the DNA methyltransferase inhibitor 5-aza-2’-deoxycitidine or the histone deacetylase (HDAC) inhibitor trichostatin A was shown to contribute directly to reprogramming of nonosteoblasts into functional osteoblasts.50,51 Cardiomyocytes were also reprogrammed epigenetically using a combination of epigenetic drugs.52 Fibroblasts were able to convert into cardiomyocytes using cardiac-specific transcription factors (Gata4, Mef2c, and Tbx5) and epigenetic remodelling proteins.53-55 Several recent reports have demonstrated the differentiation of human pluripotent stem cells into populations of nephron progenitor cells, specifically cells of the intermediate mesoderm and the metanephric mesenchyme using the transcription factors Osr1, Pax2, and Lhx1.56 Recently, Hendry et al.57 provided the first evidence of direct transcriptional reprogramming in the kidney, whereby human proximal tubule-derived renal epithelial cells (HK2) were reprogrammed back to an embryonic nonprogenitor-like state through forced expression of six factors: Six1, Six2, Osr1, Eya1, Hoxa11, and Snai2. A more efficient approach was recently developed by Vanslambrouck et al.58 using a novel inducible piggyBac transposon system, harbouring three reprogramming factors (Six1, Eya1, and Snai2), to induce reprogramming of adult kidney cells to nephron progenitor-like cells that possess differentiation capacity. Another study by Papadimou et al.59 converted human bone marrow stromal cells into renal tubule-like cells using cell-free extracts, and were shown to improve renal function in mice following kidney injury. More recently, another group described successful reprogramming of mouse and human fibroblasts into renal tubular epithelial-like cells utilising four transcription factors: Emx2, Hnf1b, Hnf4a, and Pax8.60 These induced renal epithelial cells were shown to take up albumin by endocytosis. The administration of mesenchymal stem cells (MSC) may also constitute a future form of treatment for DN. In a streptozotocin (STZ)-mouse diabetic model, the administration of MSC improved renal function in a Type 1 DN rat model as well as podocyte damage.61,62 Numerous genetic and epigenetic factors regulating kidney morphogenesis, differentiation, and maturation have been identified through decades of progress in developmental nephrology. Although nephrogenic transcription factors have been extensively studied, the mechanisms by which chromatin remodellers modulate activation or repression of transcriptional networks are not well understood. Given that the kidney has limited regeneration capacity, further investigation will be required to elucidate the roles of kidney epigenetic factors for a better understanding of the process of nephrogenesis, as well as directed differentiation or reprogramming. Several studies have also reported the use of HDAC inhibitors in rats and mice with diabetes, showing them to have renoprotective benefits. In one study, investigators reported that the broad-spectrum HDAC inhibitor trichostatin A attenuated the upregulation of both smooth muscle actin and fibronectin, and downregulation of E-cadherin.63 In another study, vorinostat was tested in STZ-diabetic rats and was found to decrease tubule cell proliferation, glomerular hypertrophy, and renal enlargement.64 Another major class of HDAC inhibitor, valproate, when injected into STZ-diabetic rats, attenuated renal fibrosis and tubule cell injury.65 Kidney organoid technology combined with CRISPR/Cas9 is providing a novel experimental platform for mechanistic studies of kidney gene function at an epigenetic level. Liu et al.66 have adapted the CRISPR/Cas9 gene-editing technology to edit DNA methylation, correlating specific modifications with chronic kidney disease. In summary, genetic factors as well as epigenetic factors play a significant role in DN.67-70
CONCLUSION
This review highlights the regulatory effect that epigenetic modifications exert in DN. Defining epigenetic signatures through the stages of kidney disease could provide novel strategies to develop cutting edge therapeutic interventions for curing the disease. It is precisely these epigenetic marks that are the obstacles for cell fate conversion. Major challenges include overcoming these epigenetic hurdles which will only be resolved once a greater understanding of the epigenome is achieved. Given that the mammalian kidney has very limited regenerative capacity, direct reprogramming together with HDAC inhibitors have emerged as promising approaches for ameliorating the disease state.