Abstract
Diabetes is an important public health concern associated with significant morbidity, premature mortality, and health-system costs. Its global prevalence has nearly doubled since 1980, rising from 4.7% to 8.5% in the adult population in 2014. Additionally, the number of diabetic adults in the world increased from 108 million in 1980 to 422 million in 2014, with the majority of people affected by Type 2 diabetes mellitus (T2DM). More common in the elderly, T2DM frequently coexists with osteoporosis, causing >8.9 million fractures annually worldwide. On the other hand, skeletal fragility has emerged as a new complication of diabetes itself. Compared with osteoporosis, T2DM reduces bone quality rather than bone mineral density. Although DM-related complications are important in the aetiology, the effects of medications on bone metabolism and fracture risk should not be neglected. Common drugs used for T2DM might have a positive, neutral, or negative impact on skeletal health. This issue has clinical significance because many T2DM patients receiving therapy are in the age range at greatest risk of bone fractures. This review focusses specifically on and summarises the skeletal effects of recently marketed glucagon-like peptide-1 receptor agonists (GLP-1 RA), dipeptidyl peptidase-4 inhibitors (DPP-4i), and sodium-glucose cotransporter 2 inhibitors (SGLT2i).
INTRODUCTION
There is growing evidence indicating the effects of medications for Type 2 diabetes mellitus (T2DM) on bone metabolism and fracture risk. This issue has clinical significance because many patients receiving anti-hyperglycaemic therapy are within the age range of greatest fracture risk. At present, there is consistent evidence for the adverse skeletal effects of thiazolidinediones in particular. Rosiglitazone-associated fractures were first reported as an adverse outcome in the ADOPT study.1 The cumulative incidence was 15.1% after 5 years of rosiglitazone treatment compared with 7.3% with metformin and 7.7% with glyburide treatment. Additionally, a case-control analysis confirmed the association of thiazolidinedione use with incident fractures, mainly in the hips and wrist,2 and this association was found to be independent of age or sex. A meta-analysis of 22 randomised controlled trials (RCTs) reported an increased fracture risk in women with rosiglitazone and pioglitazone use,3 however the risk had no clear association with duration of exposure to this class of drugs.3 In bone, peroxisome proliferator-activated receptor-γ (PPAR-γ) activation with thiazolidinediones promotes the differentiation of pluripotent mesenchymal bone marrow-derived stromal cells to adipocyte precursors, at the expense of osteoblast precursors. This leads to increased fat accumulation, unbalanced bone remodelling, and ultimately an increased fracture risk.4 However, other potential mechanisms, including an indirect negative effect of thiazolidinediones on osteoblasts via enhanced secretion of adipokines such as fibroblast growth factor (FGF)-21, adiponectin, and chemerin, remain to be elucidated in humans.
Studies on the association between older therapeutic modalities and fracture risk suggest that metformin and sulfonylureas may have either a neutral or beneficial effect on bone.1,5-8 On the other hand, the skeletal effects of insulin treatment in T2DM are controversial. Observational studies have shown in some instances an increased risk of fractures with insulin treatment1,5-10 although observational data should always be considered with caution due to prescription bias. Furthermore, there are no RCTs assessing the direct effect of insulin treatment on bone health meaning that it is difficult to draw conclusions on whether the above-mentioned association is related to a direct effect of insulin treatment, or to the associated conditions prompting the prescription of insulin (severity of DM, increased prevalence of concomitant diseases, unwanted weight loss, etc.). However, the higher incidence of insulin-associated fractures remains unchanged, even when adjusted for the duration of DM and a wide panel of confounders.5,6,10 Additionally, insulin therapy is associated with an increased frequency of severe hypoglycaemic events and an increased risk of falling, contributing to fracture risk.10,11 These findings suggest that the possible detrimental effect of insulin is not due to direct adverse effects on bone metabolism. In fact, insulin stimulates osteoblast differentiation and the synthesis of bone matrix.12
Amylin, a pancreatic β cell-derived hormone, affects bone metabolism by stimulating osteoblast proliferation and thus bone formation, and by inhibiting osteoclast-mediated bone resorption.13,14 Little evidence exists for the effect of amylin, or its analogue pramlintide, on diabetic bone. Amylin treatment has been shown to normalise bone strength, bone mineral density (BMD), and trabecular microarchitecture in studies of the streptozotocin rat.15,16 The only published study in human beings reported no interval change in lumbar spine BMD or bone turnover markers in 23 patients with T1DM without osteopenia after 1 year of pramlintide therapy.17 The implications of this fairly small uncontrolled study on bone metabolism in patients with T2DM are unclear. In recent years, three new classes of drugs have been introduced for the treatment of T2DM: glucagon-like peptide-1 receptor agonists (GLP-1 RAs), dipeptidyl peptidase-4 inhibitors (DPP-4i), and sodium-glucose cotransporter 2 inhibitors (SGLT2i). This review focusses specifically on, and summarises the skeletal effects of these recently marketed drugs (Table 1).
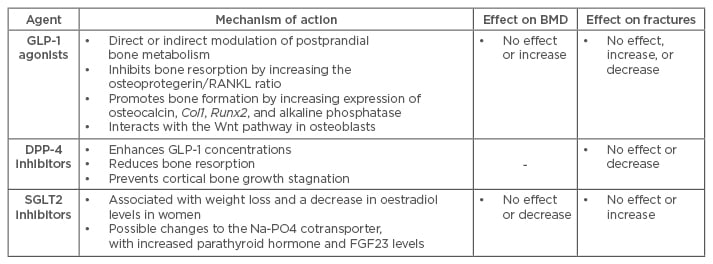
Table 1: Newest three classes of anti-hyperglycaemic drugs and their effects on bone metabolism, bone mineral density, and fractures.
BMD: bone mineral density; GLP-1: glucagon-like peptide-1; DPP-4: dipeptidyl peptidase-4; SGLT2: sodium-glucose cotransporter Type 2; RANKL: receptor activator of nuclear factor-κ B ligand; FGF23: fibroblast growth factor 23.
INCRETIN-BASED THERAPIES
Glucagon-like peptides (GLP-1, GLP-2) and glucose-dependent insulinotropic polypeptide (GIP) are gut-derived incretin hormones produced mainly in the postprandial state. They exert their actions through activation of G protein-coupled specific cell-surface receptors in different target tissues, acting on both osteoblasts and osteoclasts.18 Experimental data indicate that incretins have beneficial and protective effects on bone mass and quality. Several studies have indicated that GIP can act both as an anti-resorptive and anabolic hormone, whereas GLP-2 acts primarily as an anti-resorptive hormone.19-21 In a double-blind placebo-controlled dose-ranging trial, treatment with daily doses of 0.4, 1.6, and 3.2 mg of GLP-2 for 120 days resulted in a significant dose-dependent increase in total hip BMD in postmenopausal women.21 Additionally, GLP-1 RAs may, either directly or indirectly, shift the balance of bone homeostasis towards bone formation.22 GLP-1 stimulates proliferation of mesenchymal stem cells and inhibits their differentiation to adipocytes.23 GLP-1 receptor knockout mice present with cortical porosity due to increased osteoclast number and activity, and with reduced bone strength at anatomical and tissue levels.24,25 The decrease in bone strength at the tissue level is associated with a reduction in collagen cross-linking but not an alteration of bone mineral.25 Treatment with GLP-1RAs has increased bone mass in different models of osteopenia in rodents.26,27 GLP-1 was recently demonstrated to functionally interact with osteoblastic cells through a receptor independent of the ‘classic’ cAMP-linked GLP-1 receptor.26,28 These data suggest that activation of such specific receptors may have direct osteogenic effects, whereas GLP-1 may also act indirectly through a calcitonin dependent pathway.24 GLP-1 receptors expressed on thyroid C cells increase the secretion of calcitonin, which could contribute to the postprandial decrease in bone resorption.29
Incretins might play a role in bone homeostasis by regulating the acute interaction between nutrient ingestion and bone turnover.19,20,30 This interaction may represent the physiological adaptations of organisms to variable energy and nutrient intake.19,20,30 Energy/nutrient excess alters the remodelling process to favour bone formation and permits the organism to maximise skeletal strength. In contrast, bone remodelling is balanced to favour bone resorption during times of energy insufficiency in order to control and maintain calcium homeostasis.30 Based on available data, it is conceivable that incretin-based therapies may be a good treatment alternative in diabetics with a high risk of falls and fractures due to their bone sparing potential, and most importantly, due to their low risk of hypoglycaemia.
Glucagon-Like Peptide-1 Receptor Agonists
GLP-1 RAs mimic GLP-1 but are resistant to degradation by DPP-4. Two classes have received a market authorisation in Europe and the USA: exenatide (synthetic exendin-4) and liraglutide. Exendin-4 exhibits dual regulatory effects on bone turnover. In different animal models it was found to inhibit bone resorption by increasing osteoprotegerin/receptor activator concentrations, affecting the nuclear factor-κ B ligand ratio, while promoting bone formation by increasing the expression of osteocalcin, collagen I, runt-related transcription factor 2, and alkaline phosphatase, and by interacting with the Wnt pathway in osteoblasts.26,31,32 Exendin-4 reduced serum levels of sclerostin and increased femoral BMD in otsuka long-evans tokushima fatty (OLETF) rats, characterised by mild obesity, late onset of hyperglycaemia, hypertriglyceridaemia, insulin resistance, and impaired insulin secretion, thereby bearing resemblance to human T2DM.31
Although data from rodent models consistently suggest the presence of an osteoanabolic effect of incretins, there is little human evidence.24,26-28,31 In a small, randomised, placebo-controlled trial, a 44-week treatment with exenatide did not affect the serum levels of bone turnover markers or BMD in patients with T2DM.33 However, the relevant weight loss of 6% seen after active treatment does make the interpretation of these results problematic. Recently, Mabilleau et al.34 reported that the use of exenatide and liraglutide did not modify the fracture risk in T2DM as compared to a placebo or other anti-hyperglycaemic medications (such as glimepiride, sitagliptin, or insulin) in a meta-analysis of seven RCTs. A second meta-analysis by Su et al.35 included 16 RCTs of variable duration (12–104 weeks) exploring the association between fractures and GLP-1 RAs in general, focussing on liraglutide and exenatide in particular. Overall, the effect of GLP-1 RAs on fracture risk seemed to be neutral. Importantly, short-term GLP-1 RA use, stratified by cumulative or average daily dose, was not found to be associated with fracture risk compared with the use of other anti-hyperglycaemic drugs in a case-control study performed using Danish National Health Service data during the years 2007–2011.36 On the other hand, the fracture risk associated with different GLP-1 RAs in the meta-analysis by Su et al.35 was divergent. The use of liraglutide reduced non-vertebral fracture risk by 62% whereas exenatide doubled the fracture risk. Of note, patients taking exenatide tended to exhibit more weight loss, higher haemoglobin A1c (HbA1c) levels, and an increased incidence of hypoglycaemia (although no difference in reported falls was seen) compared with patients on liraglutide. All of these factors may have contributed to the increased fracture risk, however several methodological aspects limit the generalisation of these results: i) fractures were not the primary endpoint in RCTs included in both meta-analyses but rather serious adverse events, and ii) the number of fracture events was quite low. Additionally, no large-scale long-term trials are yet available for this class of drug.
Dipeptidyl Peptidase-4 Inhibitors
The pharmacological effect of DPP-4i is to prolong the action of GLP-1, therefore their effect on bone is assumed to be similar to that of GLP-1 RAs. In a recent preclinical study, sitagliptin use attenuated a decrease in trabecular number and an increase in trabecular spacing in diabetic rats, most likely through the reduction of bone resorption.37 Furthermore, it prevented cortical bone growth stagnation, resulting in stronger femora. All these effects were independent of glycaemic control. Human studies investigating the effect of DPP-4i on bone are scarce. Only one clinical trial has reported that vildagliptin therapy over a period of 1 year was not associated with changes in postprandial circulating levels of bone resorption markers or calcium homeostasis in drug-naïve patients with T2DM and mild hyperglycaemia, compared with baseline and placebo data.38 This neutral role of vildagliptin suggests that the fracture risk of T2DM is not altered after treatment with DPP-4i.
Results from a meta-analysis initially showed a 40% reduction in fracture incidence with DPP-4i compared with placebo or other anti-hyperglycaemic treatments.39 However, a retrospective population-based cohort study conducted by Driessen et al.40 reported no fracture risk difference comparing current users of DPP-4i to non-diabetic controls. Additionally, fracture risk was not elevated in T2DM patients after adjusting for the use of other antidiabetic drugs (metformin, sulfonylurea, thiazolidinediones, and insulin).40 Differences between these two trials can be explained by several factors. Firstly, RCTs included in the meta-analysis did not collect fractures as a primary endpoint but rather as severe adverse events. In contrast, the retrospective cohort study used data from the Clinical Practice Research Datalink (CPRD) in the UK which had identified hip fractures with high accuracy.40 Secondly, follow-up duration was different, with short-term follow-up in the meta-analysis (mean duration, 35 weeks) and a longer duration in the cohort study (median duration 5 years; actual duration 1.3 years).39,40 In addition, the limited number of events and the duration of follow-up does not exclude the possibility of a causal observation.
Driessen et al.41 also conducted a case-control study using data from the Danish National Health Service. In keeping with their previous findings, short-term use of DPP-4i in cases who sustained a fracture was not associated with risk of any fracture or major osteoporotic fracture as compared to users of other anti-hyperglycaemic drugs. Furthermore, increasing daily dose and cumulative DPP-4i exposure was not associated with fracture risk. A large-scale cardiovascular outcome trial with saxagliptin similarly failed to detect any significant difference in the number of bone fractures between saxagliptin and placebo users.42 However, this study enrolled patients with relevant comorbidities, such as renal impairment, which may have masked a potentially beneficial effect. More research is needed to elucidate whether DPP-4i are able to increase bone formation and inhibit bone resorption, and whether this effect is sufficient to reduce fracture risk in humans.
SODIUM-GLUCOSE COTRANSPORTER TYPE 2 INHIBITORS
Considering the mechanism of action, there is a concern that SGLT-2Is potentially affect bone mass and the associated fracture risk. A 50-week, double-blind, placebo-controlled study of bone outcomes with dapagliflozin and its 102-week extension did not record fractures in either the active or placebo groups.43,44 These data are however in contrast with recently published skeletal safety data, showing an increased fracture incidence associated with the use of dapagliflozin and canagliflozin.45,46 In a double-blind placebo-controlled study of adults with T2DM and moderate renal impairment, substantial increases in fracture numbers were noted in the dapagliflozin groups, with a suggestion of dose dependence.45 Of the 13 total fractures recorded during a 104-week follow-up period, none occurred in the placebo group, 5 (6.0%) fractures happened in the 5 mg group, and 8 (9.4%) occurred in the 10 mg group. All fractures occurred following trauma and were mostly of low impact. Similarly, the incidence of fractures was higher with canagliflozin (2.7%) versus non-canagliflozin (1.9%) in the overall population of patients from nine placebo and active-controlled, randomised, double-blind, Phase III studies with scheduled exposures to canagliflozin 100 or 300 mg for 1 year or longer, with the same incidence in the canagliflozin 100 and 300 mg groups.46 Fracture risk did not increase during Year 1 of treatment, but did increase during Year 2.47
Ljunggren et al.43 reported no significant changes from baseline levels of bone turnover markers and BMD after dapagliflozin treatment in patients with normal-to-mild renal impairment. On the other hand, canagliflozin has been reported to increase bone turnover.48-50 T2DM patients and overweight/obese individuals without T2DM showed modest increases in the bone resorption marker β-carboxy-terminal telopeptide of Type I collagen (β-CTX) with canagliflozin but no meaningful changes in other biomarkers in 12-week Phase II clinical studies.48,49 In a placebo-controlled Phase III clinical trial in T2DM patients aged 55–80 years, canagliflozin was associated with a dose-dependent increase in β-CTX, accompanied by increased osteocalcin levels at Week 52.50 Canagliflozin doses of 100 and 300 mg showed small but significant decreases in total hip BMD over 104 weeks. No meaningful changes in volumetric BMD or bone strength at the spine or femoral neck were observed, suggesting no impact on bone quality.50 The generation of additional data would be needed to put the association between fractures and the changes in bone biomarkers and BMD in individual patients into context. Additionally, the difference between canagliflozin and dapagliflozin may be due to compound-specific rather than mechanism-based effects on BMD or bone turnover biomarkers. It may also result from dose selection rather than any intrinsic differences. Whereas the 300 mg dose of canagliflozin delivers maximum inhibition of SGLT-2, the 10 mg dose of dapagliflozin only submaximally inhibits SGLT-2.47,51 Thus, canagliflozin 300 mg/day treatment may be more efficacious than dapagliflozin 10 mg/day with respect to all mechanism-based pharmacological actions. However, head-to-head trials are needed before firm conclusions can be drawn.
There are plausible pathophysiological mechanisms with the potential to mediate detrimental skeletal effects of SGLT-2Is. Firstly, the use of SGLT-2Is is consistently associated with weight loss.50,52 These may contribute to bone loss in part due to a direct effect of reduced soft-tissue mass on bone through reduced mechanical loading.53 Additionally, a decreased volume of fat tissue may lead to reductions in aromatase activity, which lowers the production of oestradiol and consequently increases bone turnover.54,55 The decrease in oestradiol in women with canagliflozin reported by Bilezikian et al.50 is consistent with this hypothesis. Additionally, SGLT-2 inhibition may reduce sodium transport in proximal tubule epithelial cells, leading to an increased phosphate flux through sodium-phosphate cotransporters and to increased serum phosphate.47,56-58 This effect may stimulate the secretion of parathyroid hormone (PTH), whose sustained increase enhances bone resorption and increases the risk of fractures. Along this line of enquiry, preliminary clinical data has indicated increased serum phosphate and PTH levels during dapagliflozin treatment.56,57 Ljunggren et al.43 confirmed small increases in serum phosphate concentrations with dapagliflozin however no change in serum PTH was observed.
Increased serum phosphate and PTH concentrations can induce FGF23 expression and secretion from osteoblasts and osteocytes, and increased levels have been associated with bone disease.59 In view of the controversy, future clinical studies should be performed to assess the effect of SGLT2i on FGF23 concentrations. If SGLT2 inhibition increases the concentrations of both PTH and FGF23, the net effect on 1,25-(OH)2 vitamin D (1,25-[OH]2D) cannot be predicted a priori, because PTH increases 1,25-(OH)2D levels whereas FGF23 decreases them. There was a slight decrease in 1,25-(OH)2D levels at higher doses of canagliflozin seen at Week 8, that returned to normal by Week 12 in Phase II trials.48,60
The existence of multiple homoeostatic mechanisms creates challenges in the interpretation of data whereby SGLT-2Is could affect bone health. For example, the endocrine functions of PTH and FGF23 contribute to the maintenance of phosphate balances by promoting renal phosphate excretion. Because of this negative feedback, the maximum increase in mean serum phosphate may be transient or small in magnitude. In the DIA 2001 trial, there were no consistent canagliflozin-associated changes in serum, urine calcium, or serum phosphorus concentrations,48 however there was a small, non-dose dependent increase in PTH at Week 3 in the canagliflozin treatment groups which returned to baseline by Weeks 6 and 12.48 In two other 12-week Phase II studies there were small increases from baseline levels in both urine and serum phosphorus concentrations but there was no change in serum or urine calcium.49,60
Finally, it is important to recognise that most SGLT2i-treated patients do not have bone fractures and that they may be most likely to occur in the subpopulation of outlier patients with above-average changes in bone-related parameters. The increased fracture risk with canagliflozin was driven by fractures in the CANaliflozin cardioVascular Assessment Study (CANVAS) starting within the first few weeks after study drug initiation, with a continued rate of increase thereafter.46 The cause of the increased fracture risk is unknown. CANVAS patients were older, with a prior history/risk of cardiovascular disease, lower baseline estimated glomerular filtration rates, and higher diuretic use. The small, inconsistent changes in total hip BMD (not femoral neck, lumbar spine, or distal forearm BMD) observed with canagliflozin over 104 weeks alongside an early increase in fractures that was observed only in a subgroup of patients suggest that extrinsic factors related to canagliflozin, possibly related to falls or other indirect effects of canagliflozin on bone strength, may be a more likely explanation for this observed imbalance.46,50
CONCLUSION
The effect on bone metabolism and fracture risk deserves to be included in the evaluation of risk-benefit ratios of anti-hyperglycaemic drugs. Both positive and negative expectations of newer drugs still need to be confirmed and future research should attempt to specifically identify patients who are most susceptible to the development of drug-induced bone fractures.