
Meeting Summary
Psoriasis and atopic dermatitis (AD) are both T-cell driven, chronic inflammatory skin disorders. This symposium aimed to discuss the distinct and overlapping clinical characteristics of these diseases and described how improved understanding of the immunopathological pathways involved has impacted treatment paradigms. With insight from his clinical experience, Prof Lebwohl described the key clinical and histologic features of psoriasis and AD. He also gave an overview of the evolution of systemic treatments for these diseases, which reflects growing knowledge of the T-cell driven pathologies, notably the dominance of the Th17/IL-17 pathway in psoriasis and Th2/IL-13 pathway in AD. Prof Warren provided insight into the central role of the IL-23/IL-17 axis in the immunopathogenesis of psoriasis and overviewed the registrational clinical data for approved agents targeting IL-17 and its receptor. He also discussed the importance of complete skin clearance in improving patient quality of life (QoL) and provided an update on the scope of personalised medicine in psoriasis. Prof Weidinger provided insight into the immunological pathways involved in the pathogenesis of AD and its distinct molecular profile from psoriasis, explaining the scientific rationale for, and emerging clinical data supporting, the key role of IL-13 pathways in AD.
Psoriasis and Atopic Dermatitis: Common and Distinct Clinical Characteristics
Professor Mark Lebwohl
Psoriasis and AD are chronic, systemic inflammatory diseases of the skin characterised by upregulation of proinflammatory cytokines, infiltration of immune cells to the skin, and barrier breakdown and remodelling. Commonalities between these two conditions are such that one might question whether they are distinct or represent different poles of a single spectrum.1 Psoriasis and AD are associated with distinct and overlapping symptoms which can sometimes cause confusion in terms of diagnosis. Common symptoms include itch, dry scaly skin, and redness; however, patients with psoriasis typically have clearly demarcated, chronic plaques, usually with coarse scale, whereas AD ranges from patches to thin plaques with excoriation and less defined demarcation.1 Both diseases are associated with a significant negative impact on patient QoL.2,3 There are some key differences, for example, AD usually has an early onset in childhood, whereas psoriasis generally occurs later in life. AD is often associated with comorbid allergic diseases such as asthma and/or allergic rhinitis, whereas psoriasis is commonly associated with psoriatic arthritis.1 The location of the patches and plaques can also help to differentiate between the two diseases as psoriasis is often observed on the extensor aspects of elbows and knees, whereas AD is found on the flexural aspects of the elbows and knees.1
Itch is a key symptom for both diseases. Itch was reported as one of the most severe and bothersome symptoms in a recent study evaluating a novel patient-reported psoriasis symptom diary,4 and in a recent psoriasis study, improvement in itch severity was the most important mediator of overall improvement in health-related QoL (HRQoL).5 Severity of itch, however, can potentially be used to distinguish between the two diseases as itch is more severe in AD patients which is reflected in the Hanifin and Rajka major diagnostic criteria.6 Patients with AD often have exfoliations, which is a sign of itching.
Despite the similarities between psoriasis and AD, presenting symptoms and disease history are generally sufficient to distinguish between them in the clinic. While histological features are often not needed to make a diagnosis, in some cases these can be helpful. For example, spongiotic tissue reaction patterns are commonly encountered in inflammatory dermatoses, including contact, nummular, and dyshidrotic dermatitis, and may exhibit psoriasiform hyperplasia during chronic phases. Spongiosis can be histologically characterised by the presence of microscopic vesicles in the epidermis.7 Dyshidrotic dermatitis, contact dermatitis, and volar skin psoriasis may appear virtually identical; however, volar psoriasis usually has more mounds of parakeratosis with neutrophils. Other conditions described as ‘psoriasiform dermatitis’ include: pityriasis rubra pilaris, an idiopathic condition which presents with follicle-based papules, plaques, and as palmoplantar keratoderma; and lichen simplex chronicus which is associated with intense pruritus and hyperkeratotic erythematous patches.8 Abramovits et al. reported on the occurrence of overlapping psoriasis and AD symptoms in their clinic and proposed an overlapping dermatologic entity called ‘PsEma’.9
Distinct and overlapping pathogenic mechanisms are also evident between psoriasis and AD which drive the treatment approaches available today. The first systemic treatments were the nonspecific, broad-acting immunomodulators methotrexate and cyclosporin, which provided symptomatic control.10-13 In recent years, however, systemic treatments have evolved considerably, mirroring our growing understanding of the T-cell driven pathologies of these diseases, including the dominance of the Th17/IL-17 pathway in psoriasis and Th2/IL-13 pathway in AD. Targeted biologic agents offer advantages over broad-acting immunomodulators, particularly in terms of safety profiles.14 The evolution of targeted treatment for psoriasis started with approval of monoclonal antibodies against TNF-α, followed by approval of more targeted systemic agents that inhibit IL-12/23, IL-23, IL-17, and the IL-17 receptor subunit A (IL-17RA).15 In contrast, targeted treatment for AD has only recently become available with the approval of monoclonal antibodies against IL-4/13, with targeted treatments against IL-13 currently in development.16,17
Personalised Therapy in Psoriasis with a Focus on IL-17
Professor Richard Warren
The interplay between environmental and genetic factors is implicated in the initiation of psoriasis and the activation of the psoriatic inflammatory cascade. There are three immune axes involved in this cascade, all of which are mediated by different subsets of T cells: Th1 cells which secrete TNF-α and interferon (IFN)-γ, Th22 cells which secrete IL-22, and Th17 cells which secrete IL-17.18,19 All three T cell subtypes play an important role in the immunopathogenesis of psoriasis; however, Th17 cells have been shown to be the main driver of the disease phenotype. Activated dendritic cells release IL-23 which stimulates Th17 cells to produce IL-17 family cytokines. In psoriasis, expression of IL-17A, IL-17C, and IL-17F is increased; these cytokines activate IL-17RA complexes on keratinocytes and immune cells leading to inflammation and the clinical manifestations of the disease.20-24 As multiple cell types can produce IL-17, inhibiting upstream cytokines such as TNF-α, IL-12, or IL-23 only partly attenuates IL-17 production.25 This leaves other cytokines to synergise with residual levels of IL-17 contributing to the inflammatory response.26,27 Inhibition of IL-17RA, however, directly blocks the action of multiple IL-17 cytokines (specifically IL-17A, IL-17C, IL-17E, IL-17F, and IL-17A/F). This rapidly and completely normalises the inflammatory response suggesting that IL-17RA plays a key role in the pathogenesis of psoriasis.20,21,27
Due to the importance of the IL-23/17 axis, targeting IL-23 or IL-17 should have great therapeutic potential in psoriasis.28 Treatment of psoriasis has evolved considerably over recent years with the introduction of highly efficacious IL-17 and IL-23 inhibitors. To date, three IL-17 inhibitors are approved for moderate-to-severe plaque psoriasis: secukinumab and ixekizumab which selectively bind to IL-17A, and brodalumab which selectively binds to IL-17RA and thereby blocks activity of IL-17A, IL-17C, IL-17E, IL-17F, and IL-17A/F molecules (Figure 1).29
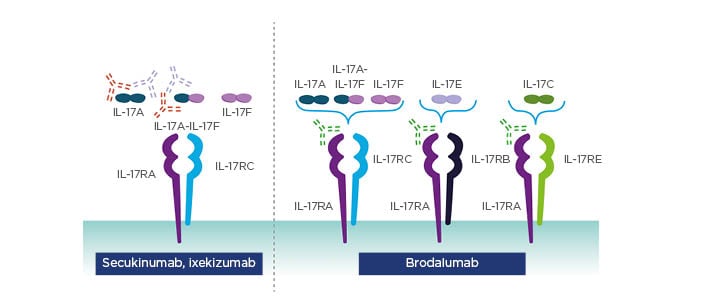
Figure 1: Approved IL-17 inhibitors target the cytokine or the receptor.
Adapted from Patel et al.29
Reproduced from Effect of IL-17A blockade with secukinumab in autoimmune diseases. Patel D et al. Ann Rheum Dis. 72 (Suppl 2):ii116–23, 2013 with permission from BMJ Publishing Group Ltd.
Approved Agents Targeting IL-17
Secukinumab was the first IL-17-specific biologic approved for psoriasis based on data from two registrational Phase III trials, ERASURE and FIXTURE.30 Together, these studies investigated secukinumab (150 mg and 300 mg doses every 4 weeks [Q4W]) versus placebo and the TNF-α blocker, etanercept in 2,044 patients with refractory, moderate-to-severe plaque psoriasis.
At Week 12, secukinumab was superior to placebo and etanercept for the co-primary efficacy endpoints of Psoriasis Area and Severity Index (PASI) 75 (p<0.001) and response of 0 or 1 on the modified Investigator’s Global Assessment (IGA; p<0.001), and was also superior for other efficacy endpoints including PASI 90 and 100 at Week 12. The benefit with secukinumab was dose-dependent, with higher levels of response observed with the higher dose of 300 mg.30 The SCULPTURE extension study demonstrated that PASI responses (PASI 75, 90, and 100) with secukinumab can be maintained for up to 5 years (observed analyses; small reductions were observed with multiple imputation and last observation carried forward analyses).31 In the ERASURE and FIXTURE studies, the incidence of serious adverse events (AE) with secukinumab was low and comparable to placebo. The most common AE observed with secukinumab were nasopharyngitis, headache, and diarrhoea. There was a slightly higher incidence of candida infections in patients receiving secukinumab but this did not lead to discontinuation.30 Crohn’s disease and ulcerative colitis are other AE of special interest as IL-17 inhibitors were found to be ineffective for the treatment of these diseases and there were concerns that IL-17 treatment may exacerbate inflammatory bowel disease (IBD) in psoriasis patients; however, the incidence of IBD with secukinumab was low.30 The long-term safety profile at 5 years was similar to the induction phase and no new safety signals were observed.31
European approval of ixekizumab for moderate-to-severe plaque psoriasis in 2017 was based on data from the UNCOVER-1, 2, and 3 Phase III studies. UNCOVER-2 and 3 assessed ixekizumab 80 mg every 2 weeks (Q2W) and Q4W versus placebo and etanercept in 1,224 and 1,346 patients, respectively. All patients treated with ixekizumab received an induction dose of ixekizumab 160 mg followed by 80 mg Q2W up to Week 12 followed by 80 mg Q2W or Q4W.32 At Week 12, pooled data from these studies showed that ixekizumab was superior to etanercept and placebo for the co-primary endpoints of PASI 75 (p<0.001) and proportion of patients achieving static Physician’s Global Assessment (sPGA) of 0 or 1 (p<0.001). This benefit was also observed with ixekizumab for PASI 90 and 100.32 Among patients who received the recommended dose of ixekizumab (160 mg at Week 0, ixekizumab 80 mg Q2W up to Week 12, and Q4W thereafter) in the long-term extension of UNCOVER-3, clinical response was maintained up to Week 156. Long-term PASI response was assessed using observed and multiple imputation analyses, whereas long-term sPGA response was analysed using a modified non-responder imputation in which patients who discontinued due to AE, lack of efficacy, or relapse were considered non-responders, and as such, data was imputed as non-responder imputation.33 The safety profile of ixekizumab across the three pivotal UNCOVER trials was manageable and the most common AE were nasopharyngitis and injection site reactions. A dose-dependent incidence of candida infections and low incidence of IBD were observed.32 A similar safety profile was observed in the long-term extension at Week 156.33
Approved Agent Targeting the IL-17 Receptor
AMAGINE-1, 2, and 3 were the pivotal Phase III trials leading to approval of brodalumab: a human anti-IL-17RA monoclonal antibody. AMAGINE-2 and 3 were placebo-controlled, active comparator studies which recruited a total of 3,712 patients.34Patients received ustekinumab (anti-IL-12/23), placebo, or brodalumab (140 mg Q2W or 210 mg Q2W) during the 12-week induction phase. Individuals who received brodalumab induction were then re-randomised to receive brodalumab 210 mg Q2W or brodalumab 140 mg Q2W, Q4W, or Q8W during the 40-week maintenance phase. Patients randomised to placebo induction received maintenance brodalumab 210 mg Q2W. Individuals randomised to ustekinumab induction also received ustekinumab as maintenance therapy, and from Week 52 could receive brodalumab 210 mg Q2W in an open-label extension study.34
In both studies, at Week 12, the proportion of patients with PASI 75 (AMAGINE-2: p=0.08; AMAGINE-3: p=0.007), sPGA 0 or 1 score (AMAGINE-2 and -3: p<0.001), and PASI 100 (AMAGINE-2 and -3: p<0.001) was greater for brodalumab 210 mg Q2W versus ustekinumab.34 Response to continuous brodalumab 210 mg Q2W was maintained throughout 120 weeks of treatment in AMAGINE-2 (using as observed analysis). Furthermore, switching from ustekinumab to brodalumab 210 mg Q2W at Week 52 resulted in improved outcomes, with similar levels of skin clearance response being achieved at Week 120 compared with patients who received brodalumab 210 mg Q2W from the start of the study.35 In a pooled analysis of AMAGINE-2 and 3, brodalumab was associated with high rates of complete clearance over time (Week 0–52) compared with ustekinumab, regardless of prior therapy (systemic/biologic naïve, systemic treated, biologic treated with or without failure).36 In AMAGINE-2 and 3, the incidence of AE was low and comparable to ustekinumab and placebo, and the most common AE were nasopharyngitis, upper respiratory tract infection, headache, and arthralgia. There was a low rate of candida infection, but no incidences of Crohn’s disease were observed.34 Long-term data demonstrated a similar safety profile, consistent with the induction phase, and no new safety signals.35
Given there are several biologics approved for moderate-to-severe psoriasis, comparing agents can be challenging due to limited head-to-head data to inform treatment decisions. Network meta-analyses (NMA) can be used to compare treatments using common comparators and are important in informing payors of differences between agents; however, they can include data from patients treated with unlicensed doses and analysis methodologies can vary across different NMA. A recent NMA compared the efficacy of available biologic and non-biologic systemic therapies for patients with moderate-to-severe plaque psoriasis, analysing data from 77 trials involving 34,816 patients.37 This large-scale analysis revealed IL-17-targeted agents were among the top performing drugs, with brodalumab and ixekizumab outperforming secukinumab in terms of PASI responses and demonstrating similar efficacy to the IL-23 inhibitor risankizumab.37 It should be noted however, that NMA provide only indirect evidence and may be influenced by the methodology employed; randomised comparator trials are needed to robustly compare the efficacy of the IL-17-targeted agents both with themselves and other agents.
Optimising Treatment for Personalised Care
An important aspect of personalising care in order to optimise treatment is consideration of the patient’s perspective of their own treatment goals. When using a ‘treat-to-target’ approach, several clinical measures should be incorporated: efficacy, safety, tolerability, and patient-reported outcomes (PRO).38 PRO are particularly important because achieving complete clearance (PASI 100 or sPGA 0) is associated with improved HRQoL in the clinical trial setting and in real-world clinical practice.39,40 Caution should be exercised when reviewing these data as not all patients achieving complete clearance experience this benefit in HRQoL and this may be due to the insensitivity of the PRO used or a residual impact on patients’ wellbeing.
Predictors of Response to Biologic Treatment
Despite the availability of several effective biologic treatments, some patients are still unable to achieve an adequate response; therefore, a better understanding of the predictors of response may be beneficial. Studies from the Psoriasis Stratification to Optimise Relevant Therapy (PSORT) study group have demonstrated that serum drug levels of adalimumab and ustekinumab taken around Week 4 may be predictive of PASI 75 response at 6 months.41,42 Genetic biomarkers such as HLA-C*06:02, a susceptibility allele for psoriasis, may also be important when predicting differing responses to adalimumab and ustekinumab.43
The future of personalised care in psoriasis will integrate ‘multi-omic’ data, including drug levels, genetic biomarkers, and the proteome, to allow clinicians to predict response to treatment and effectively manage care. These approaches to personalised care are highly relevant to AD as it is a very heterogeneous disease with several underlying immunological pathways, including some involvement of IL-17.1
Atopic Dermatitis: A Th2 Dominant Disease with Greater Molecular Heterogeneity
Professor Stephan Weidinger
AD is a common inflammatory skin disease associated with a high disease burden, including a negative impact on mental health and QoL, frequent sleep disturbances, infections, and comorbidities such as asthma, rhinitis, and food allergies.44,45 AD is triggered by a complex immune pathophysiology pathway in which epidermal barrier dysfunction and Type 2 inflammation are thought to play an important role.
In AD, decreased expression of epidermal barrier proteins, including filaggrin and hornerin, contribute to skin barrier dysfunction which affects the composition of the skin microbiome, including decreased bacterial diversity and distinct bacterial colonisation patterns such as increase in the presence of Staphylococcus aureus.46,47 Filaggrin deficiency has also been found to increase epidermal permeability in animal models, which may contribute to enhanced antigen penetration and sensitisation.48,49 Epidermal barrier disruption promotes inflammation by stimulating keratinocytes to release skin alarmins such as thymic stromal lymphopoietin, IL-33, and IL-25.50,51 These mediators activate skin-resident Group 2 innate lymphoid cells to stimulate secretion of IL-13 and IL-5 which activate and recruit further immune cells to the skin, creating a positive feedback loop.50,52 Th2 lymphocytes are also activated leading to the secretion of IL-13 and IL-4.50
IL-13 is a Key Driver of Chronic Inflammation in Atopic Dermatitis Skin
While AD and psoriasis share several clinical features and pathogenic mechanisms, they are molecularly distinct diseases. A recent study of messenger RNA (mRNA) levels in AD and psoriatic skin lesions revealed that, despite common dysregulated genes, the AD genetic skin signature is dominated by Th2 cytokines (IL-13 being the most distinctive marker), while psoriasis is dominated by the IL-17 family of cytokines. Furthermore, IL13 mRNA expression was detected in all AD lesional skin samples analysed in this study, whereas IL4 mRNA expression was only detected in 40% of samples, suggesting that AD is predominantly IL-13 driven. The authors also reported a positive correlation between IL13 mRNA expression and disease severity in AD patients.53
Data presented by Prof Weidinger at EADV 2019 further supported the key role of IL-13 in driving the pathogenesis of AD. IL13 mRNA expression in in vitro skin cultures from AD patients was shown to positively correlate with the expression of key inflammatory chemokines and negatively correlate with skin barrier biomarkers;54 furthermore, addition of an IL-13 inhibitor to these skin cultures restored the expression of genes central to epithelial barrier function, including filaggrin.54 Together, these findings support the concept that IL-13 is the dominant cytokine in AD, with downstream effects on epithelial barrier function, susceptibility to infection, inflammation, itch, and skin thickening (Figure 2).52
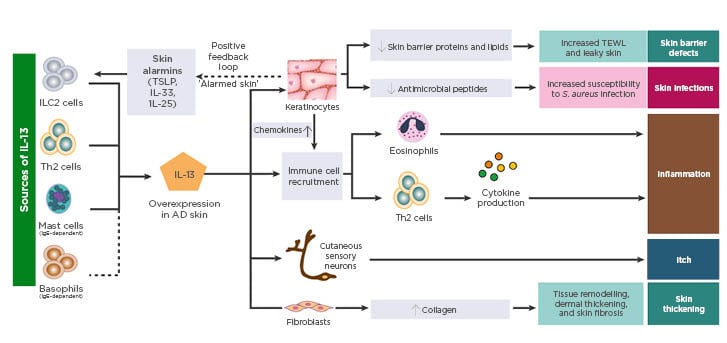
Figure 2: Sources and impact of IL-13 in atopic dermatitis skin.
AD: atopic dermatitis; ILC: innate lymphoid cells; S. aureus: Staphylococcus aureus; TEWL: transepidermal water loss; TSLP: thymic stromal lymphopoietin.
Adapted from Bieber T.52
Image included with permission from EAACI and John Wiley and Sons A/S ©2019.
Targeting IL-13 Pathways
Dupilumab is the first licensed biologic for AD and elicits its effects by binding to the shared IL-4Rα subunit of the Type I and Type II receptors blocking both IL-4 and IL-13-mediated signalling.16 Approval of dupilumab was based on two Phase III monotherapy trials (SOLO 1 and SOLO 2 [N=1,379]) and a Phase III trial investigating concomitant administration with topical corticosteroids ([TCS] CHRONOS). Pooled Week 16 monotherapy data from SOLO 1 and 2 showed that dupilumab 300 mg Q2W resulted in a greater proportion of patients achieving IGA response (score 0 or 1 and ≥2 point reduction from baseline) versus placebo (37.0% versus 9.3%; p<0.0001) and a greater proportion of patients with Eczema Area and Severity Index (EASI)-75 (47.7% versus 13.3%; p<0.0001).55 Similar outcomes were observed with dupilumab 300 mg Q2W in combination with TCS at Week 16 in the 917 patients enrolled in the CHRONOS study (IGA response: 38.7% versus 12.4%, p<0.0001; EASI-75: 68.9% versus 23.2%, p<0.0001).56 Real-world data from the TREATGermany registry indicate that dupilumab is well-tolerated in routine clinical practice, with conjunctivitis being the most frequently reported AE.57
Specific inhibition of IL-13 is currently under investigation in AD, and data from Phase IIb studies of tralokinumab and lebrikizumab have recently been reported.58,59 Tralokinumab binds to the IL-13 cytokine at an epitope overlapping the IL-13Rα receptor binding site, preventing IL-13 from binding to the IL-13Rα1 subunit of the Type II receptor.52 At Week 12 of the placebo-controlled Phase IIb study, tralokinumab 300 mg Q2W in combination with TCS was associated with a greater proportion of patients achieving IGA response (26.7% versus 11.8%; p=0.06) and EASI-75 (42.5% versus 15.5%; p=0.003) compared with placebo plus TCS. A low level of serious AE was reported, and the most common treatment-emergent AE were upper respiratory tract infection and headache.58 Lebrikizumab binds to the IL-13 cytokine at an epitope overlapping the IL-4Rα receptor binding site to prevent IL-4Rα/IL-13Rα1 heterodimerisation of the Type II receptor.52 At Week 16 of a placebo-controlled Phase IIb study, a higher proportion of patients treated with lebrikizumab 250 mg Q2W monotherapy achieved an IGA response (44.6% versus 15.3%; p=0.0023) and EASI-75 (60.6% versus 24.3%; p=0.0005) compared with placebo. The most frequent AE reported were upper respiratory tract infection and nasopharyngitis.59 Efficacy and safety data from Phase III trials for tralokinumab and lebrikizumab are required to further inform the potential utility of IL-13-specific inhibition as a therapeutic approach for AD.





