Meeting Summary
As part of the 31st European Academy of Dermatology and Venereology (EADV) Annual Congress held in Milan, Italy, and online, 7th–10th September 2022, five poster presentations outlined results from the Phase IIb study of remibrutinib (LOU064) in patients with chronic spontaneous urticaria inadequately controlled with H1-antihistamines. These posters presented data on quality of life, the need for antihistamine rescue medication to address symptoms, the time to complete urticaria control, and the safety profile of remibrutinib. The findings were outlined in these poster presentations and are summarised here.
Background
Chronic spontaneous urticaria (CSU) is characterised by the spontaneous occurrence of wheals (hives) and/or angioedema for ≥6 weeks.1 CSU can persist for several years and is associated with severe pruritus.1 Skin mast cell activation in CSU occurs via Type I autoallergy driven by IgE to autoallergens, and a Type IIb autoimmune response due to mast cell-targeted autoantibodies against IgE or high-affinity IgE receptor.2
International clinical guidelines published in 2022 state that the goal of urticaria treatment is to achieve complete symptom control safely and effectively, with normalisation of quality of life (QoL).3 However, real-world evidence from the ASSURE-CSU study,4 the AWARE study,5 and the 2-year follow-up of the AWARE study6 confirmed that in many patients CSU remains uncontrolled, undertreated, and associated with a high healthcare resource use burden, with a detrimental effect on QoL, work, sleep, and other activities.4-6
Second-generation, non-sedating H1-antihistamines at approved doses are currently recommended as first-line therapy for all types of urticaria.3 However, up to 60% of patients do not respond adequately within 2–4 weeks of starting treatment with H1-antihistamines and require increased doses (up to four-fold of the licensed dose) as second-line therapy before other treatments are considered.3,7 For many patients, the current treatments inadequately control the symptoms that impact their QoL.7-9 Hence, there is a considerable unmet need for effective and safe oral therapies for CSU with novel mechanisms of action.10
Bruton’s tyrosine kinase (BTK) is essential for signalling through the high-affinity IgE receptor receptor in mast cells and basophils.11 Therefore, BTK is an attractive therapeutic target for CSU.11 BTK inhibitors have potential efficacy in both Type I and Type IIb CSU due to BTK-mediated degranulation in mast cells and inhibition of autoantibody production in B cells.12
A Phase IIb Dose-Finding Evaluation of Remibrutinib (LOU064)
Remibrutinib (LOU064) is a novel, highly selective, and potent covalent oral BTK inhibitor, recently evaluated in a Phase IIb, first-in-patient, dose-finding, multicentre, randomised, double-blind, placebo-controlled study.13,14 This trial was conducted at 82 sites in 17 countries for adult patients with moderate-to-severe CSU for ≥6 months, and itch and hives for ≥6 consecutive weeks, despite treatment with H1-antihistamines.13 Eligible patients were required to have a Urticaria Activity Score (UAS7) of ≥16 points, and a Hives Severity Score (HSS7) of ≥8 points at baseline.13 The primary endpoint was the change from baseline in UAS7 at Week 4, with secondary endpoints evaluating change from baseline in UAS7 at Week 12 and over time, complete absence of hives and itch, well-controlled disease response, and safety and tolerability.13
A total of 311 patients were randomised to receive remibrutinib 10 mg once daily (QD), 35 mg QD, 100 mg QD, 10 mg twice daily (BID), 25 mg BID, 100 mg BID, or placebo (Figure 1).13 Approximately 90% of patients completed a 12-week treatment course in the core study, and eligible patients from this study (weekly UAS7 ≥16) had the option to roll-over into an open-label extension study at Week 12 or Week 16, where patients were treated with 100 mg remibrutinib BID for 52 weeks.13,15 All patients received second-generation H1-antihista mines at a locally approved licensed dose as background therapy throughout the study.13
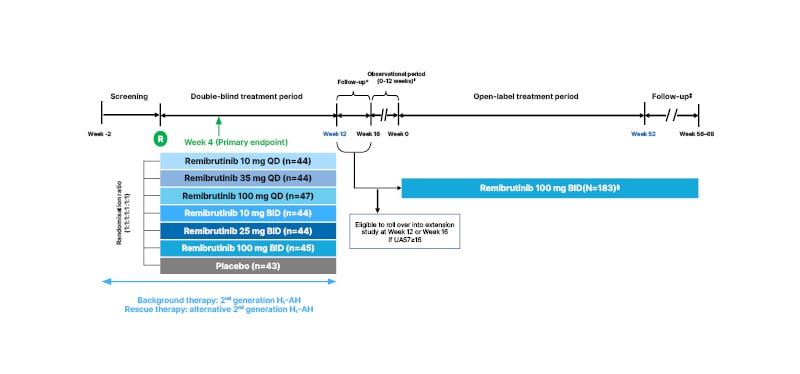
Figure 1: A dose-finding, multicentre, randomised, double-blind, placebo-controlled Phase IIb and open-label extension study in patients with chronic spontaneous urticaria.13-20
*Eligible patients (UAS7≥16) rolled over into the extension study at Week 12 or at Week 16.
†If UAS7<16 at Week 16, patients were allocated to the observational period of the extension study for up to 12 weeks. After a relapse in the extension study (UAS7≥16 at least once), the observational period was terminated, and the patient could enter the treatment period.
‡Minimum duration of the follow-up period was 4 weeks for all patients who stopped treatment with remibrutinib. Patients who achieved UAS7≤6 at Week 52 of the treatment period extended their follow-up period until relapse (UAS7≥16). Follow-up ended at Week 68 for all patients.
§Data for 183 patients were available during interim analysis (July 2021).
Background therapy was a second-generation H1-AH at a locally approved licensed posology that had to be administered daily with a stable treatment regimen throughout the study. Rescue therapy was a second-generation H1-AH at a locally approved licensed posology that differed from the background H1-AH, was eliminated primarily via renal excretion, and could only be given to treat unbearable symptoms (itch) of CSU on a day-to-day basis.
BID: twice a day; CSU: chronic spontaneous urticaria; H1-AH: H1-antihistamines; QD: once daily; UAS7: weekly Urticaria Activity Score.
Baseline demographic and disease characteristics were similar across groups.13 The mean (±standard deviation) age was 45.0 years (±14.9 years), and 71.4% of patients were female.13 The mean UAS7 score at baseline was 29.6 (±7.1).13 UAS7 scores significantly improved at Week 4 in all remibrutinib doses (p<0.0001 versus placebo), the primary endpoint.13 A rapid and significant improvement in UAS7 was observed as early as Week 1, which was maintained up to Week 12 in all doses compared with placebo.13 More patients who received remibrutinib (all doses) achieved a complete absence of hives and itch (UAS7=0) and a well-controlled disease response (UAS7≤6) over the 12-week treatment period.13 From Week 2, these responses were maintained (UAS7≤6) or gradually increased (UAS7=0) up to Week 12.13 Remibrutinib demonstrated a favourable safety profile across the dose range, with no dose-dependent pattern of adverse events (AE).13 The most frequent AEs were headache and nasopharyngitis (9.7% and 8.6% all-dose remibrutinib versus 14.3% and 7.1% placebo, respectively).13
In the Phase IIb trial, all remibrutinib doses provided clinically meaningful improvements versus placebo regarding the proportion of patients achieving UAS7=0 and UAS7≤6 over the entire treatment period up to Week 12, with a fast onset of action starting as early as Week 1.13 Safety and tolerability were favourable across the whole dose range, with no dose-dependent pattern of events.13 Remibrutinib was shown to be a promising new oral treatment option for patients with moderate-to-severe CSU.13
The following poster presentations at EADV 2022 further outlined the benefits of remibrutinib from the Phase IIb dose-finding study, specifically the time to complete urticaria control, a reduced need for rescue medication, a favourable safety profile, and improved QoL for patients.16-20
Faster Time to Complete Urticaria Control Compared to Placebo
Marcus Maurer
The current analysis examined the time to first weekly UAS7 responses in patients with moderate-to-severe CSU from the Phase IIb study.16 The median times to UAS7=0 (complete absence of hives and itch), first UAS7≤6 (well-controlled disease), and the response rate for achieving UAS7=0 and UAS7≤6 over time up to Week 12 were included.16
The median time to first UAS7=0 was shortest with remibrutinib 25 mg BID (4 weeks), compared with 100 mg BID (11 weeks) and 35 mg QD (12 weeks), and was not estimable for placebo.16 Similarly, the median time to first UAS7≤6 was shortest with remibrutinib 25 mg BID (2 weeks) compared with all other doses and was not estimable for placebo.16
As early as Week 2, up to 32.6% of patients (in the 25 mg BID arm) reached complete control (UAS7=0) compared with 0% in the placebo arm.16 By Week 12, 41.9% of patients (in the 25 mg BID arm) and 26.7–31.8% of patients on other doses of remibrutinib had reached UAS7=0, compared with 14.3% in the placebo arm.16 By Week 1, 27.9% patients (in the 25 mg BID arm) had achieved well-controlled disease (UAS7≤6), compared with 0% in the placebo arm.16 By Week 12, 55.8% (in the 25 mg BID arm) and 38.3–52.3% of patients on other doses of remibrutinib had reached UAS7≤6, compared with 28.6% of patients on placebo.16
The results demonstrated that complete response (UAS7=0) and well-controlled disease (UAS7≤6) were achieved more rapidly and by more patients with remibrutinib compared with placebo.16
A Reduced Need for Rescue Medication
Marcus Maurer
This analysis reported the use of second-generation H1-antihistamines as rescue medication in patients with moderate-to-severe CSU.17 The number of rescue H1-antihistamine tablets used over the preceding 24 hours to control itch or hives was evaluated from baseline to Week 12.17 Rescue medication allowed was second-generation H1-antihistamines eliminated mainly by renal excretion.17 The rescue medication had to be different from the standard H1-antihistamines and was given as needed to treat severe symptoms during the screening, treatment, and follow-up periods.17 The weekly use of rescue medication was calculated as the sum of the doses per day, over 7 days.17
Remibrutinib reduced the need for rescue medication as early as Week 1 compared with baseline and placebo across all doses over 12 weeks in patients with CSU (Figure 2), which was accompanied by an improvement in CSU symptoms, despite reduced use of H1-antihistamines.17 The mean weekly use of rescue medication tablets was numerically lower across all remibritunib arms compared to baseline and placebo.17 At Week 4, the use of rescue medication with remibrutinib ranged from 2.9–7.7, compared with 12.3 for placebo; and at Week 12 the use of rescue medication with remibrutinib ranged from 3.9–7.1, compared with 11.9 for placebo.17
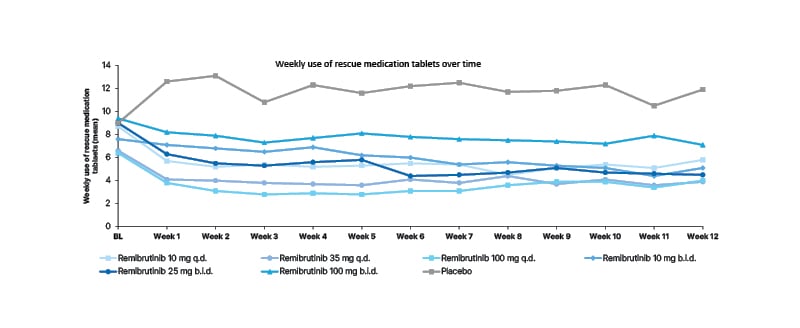
Figure 2: Reduction in weekly use of rescue medication was observed early in all remibrutinib arms and remained low throughout the study.
Full analysis set.
BID: twice a day; BL: baseline; QD: once daily.
Results demonstrated a rapid reduction in the need for rescue medication following treatment with remibrutinib across all doses, which was maintained over time in patients with CSU.17
Improved Quality of Life with Remibrutinib (LOU064)
Marcus Maurer
It is known that CSU can, and often does, have a substantial negative effect on patients’ QoL.4 The current analysis explored the changes in QoL over time in patients with CSU treated with remibrutinib compared with placebo using the Dermatology Life Quality Index (DLQI). Changes in DLQI score from baseline to Week 4 and Week 12, as well as proportions of patients achieving DLQI=0–1, were analysed at Week 4 and Week 12.18
The mean (±standard deviation) baseline DLQI score across all 311 randomised patients was 12.8 (±6.9), and ranged from 14.9 (±7.1)–10.8 (±6.7) across the group of patients that received remibrutinib, compared with 13.4 (±7.9) in the placebo group. Improvements in DLQI scores from baseline to Week 4 and Week 12 were numerically greater in all remibrutinib-treated groups than placebo. At Week 4, a decrease of 6.2–9.6 was observed with remibrutinib compared with –3.3 with placebo, and at Week 12 a decrease of 6.3–9.0 was observed with remibrutinib, compared with –4.4 with placebo (Figure 3).18
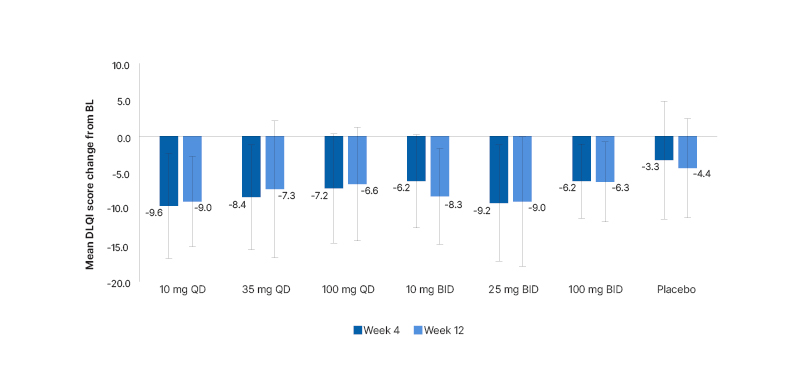
Figure 3: Changes in Dermatology Life Quality Index scores from baseline to Week 4 and Week 12.
A decrease in DLQI score indicates an improvement in QoL.
BID: twice a day; BL: baseline; DLQI: Dermatology Life Quality Index; QD: once daily; QoL: quality of life.
Further, a greater proportion of patients achieved DLQI=0–1 with remibrutinib than with placebo.18 At Week 4, a total of 29.5–51.2% of patients achieved DLQI=0–1 with remibrutinib compared with 16.7% of patients with placebo, and at Week 12 a total of 34.1–53.5% of patients achieved DLQI=0–1 with remibrutinib compared with 28.6% of patients with placebo.18 The group that contained the highest proportion of patients to achieve DLQI=0–1 was the group of patients who received remibrutinib at a dose of 25 mg BID (51.2% and 53.5% at Weeks 4 and 12, respectively).18
All remibrutinib doses, and particularly 25 mg BID, provided marked improvements in QoL of patients with CSU as early as Week 4 up to Week 12 compared to placebo.18 These findings were consistent with previously reported results of remibrutinib treatment on weekly UAS7.13
A Favourable Safety Profile Across All Doses of Remibrutinib
Ana Giménez-Arnau, Martin Metz
These two analyses reported the safety and tolerability of remibrutinib for up to 52 weeks in patients with CSU from the final analysis of the dose-finding Phase IIb trial and an interim analysis of its open-label extension period.19,20 A total of 183 patients were recruited from the core study into the extension study, and at the time of interim analysis, 65 out of 183 patients (36%) had completed the full 52-week treatment in the extension period.19,20 Patient demographics (age, gender, and weight) were similar across studies.19,20 Safety assessments comprised AEs, serious AEs, AEs of special interest, infections and infestations, vital signs, ECG, and laboratory parameters.19,20
Analyses showed that safety and tolerability of 52-week treatment with remibrutinib 100 mg BID were comparable to any remibrutinib dose in the core study.19,20 No new safety signals were observed with longer-term exposure to remibrutinib in patients with CSU.19 Haemorrhages were infrequent, minor, and non-serious cutaneous and mucosal events, primarily petechiae and purpura.19
The analysis of laboratory parameters, vital signs, and ECG findings did not reveal any significant safety concerns.19 Notably, liver function tests identified only isolated cases of increased alanine aminotransferase (ALT).19 One patient in the extension study had more than three-times the upper limit of normal for ALT, which normalised within 4 weeks (this patient withdrew from the study for personal reasons).19 One patient in the core study underwent a transient increase in ALT of more than five-times the upper limit of normal, which resolved during treatment.19
The rate of infections and infestations was comparable between remibrutinib (all doses) and placebo and did not increase with longer-term exposure to remibrutinib.19,20 Most infections were of unspecified pathogen, of which upper respiratory tract infection was the most common.20 Rates of viral infections were low, with primarily coronavirus ones reported, reflecting the impact of the COVID-19 pandemic.20 Bacterial and fungal infections were rare.20
The final analysis of the dosing in the Phase IIb trial and an interim analysis of the extension study showed a favourable safety profile and good tolerability of remibrutinib in patients with CSU, including long-term treatment with an oral dose of 100 mg BID remibrutinib for up to 52 weeks.19,20
Conclusions
The Phase IIb dose-finding study of remibrutinib (LOU064), a novel, highly selective, and potent covalent oral BTK inhibitor, demonstrated significant improvements in disease activity as measured by the UAS7 in patients with CSU at Week 4 and Week 12 when compared with placebo. Clinical efficacy was maintained throughout the treatment, with a rapid clinical response, and a favourable safety profile across the entire dose range, with no dose-dependent events. Further analyses of the Phase IIb dose-finding study of remibrutinib demonstrated complete response and well-controlled disease. Also, there was a reduced need for rescue medication and reduced CSU symptoms despite the reduced use of H1-antihistamines. Significant improvements in the QoL of patients with CSU were present as early as Week 4 and up to Week 12. A favourable safety profile and good tolerability of remibrutinib were found, including with long-term treatment of up to 52 weeks. These results support that remibrutinib is a promising new oral treatment option for patients with CSU.
Remibrutinib (LOU064) is currently in Phase III development to evaluate the efficacy and safety of remibrutinib for the treatment of CSU in adults inadequately controlled by second-generation H1-antihistamines. REMIX-121 and REMIX-222 are 52-week, global, multicentre, randomised, double-blind, parallel-group, placebo-controlled studies. BISCUIT23 is a multicentre, open-label, single-arm study to investigate the safety, tolerability, and efficacy of remibrutinib in a Japanese population. The results of these clinical trials are awaited.





