Abstract
Congenital ichthyosis represents keratinisation disorders characterised by abnormal skin scaling across the entire body, leading to a red, denuded, and scaly appearance. A subgroup of this is the autosomal recessive congenital ichthyosis (ARCI), characterised by a severe phenotype and classified according to the molecular mechanisms that underlie the disease. This article reports on the cases of two female patients with symptoms of palmoplantar keratoderma since birth and a variant in the ABCA12 gene that encodes an amino acid glucosylceramide transporter known as ABCA12. The primary role of ABCA12 is to facilitate the transport of molecules across cell and intracellular membranes. Variants involve large deletions and nonsense variants, resulting in a truncated protein that contributes to the severity of harlequin ichthyosis. However, the patients reported in this article present an attenuated phenotype with palmoplantar keratoderma. The subdued presentation in these patients might be elucidated by their compound heterozygous status.
Key Points
1. Most cases described in relation to the ABCA12 gene are associated with harlequin ichthyosis, a lethal phenotype that appears in neonatal stages. Generally, these cases are associated with homozygous variants. In cases of compound heterozygosity, milder phenotypes occur, such as palmoplantar keratoderma, seen in this case report, highlighting the genotype-phenotype activation of a spectrum of disease.2. This article presents a case report of two sisters with symptoms of palmoplantar keratoderma. In one of them, the exome identified a pathogenic nonsense variant and a probable pathogenic intronic variant in the ABCA12 gene, in a compound heterozygous state. A literature review was carried out, identifying that this gene has a higher phenotypic frequency of harlequin ichthyosis, and is generally due to homozygous variants. In contrast, biallelic missense variants mainly lead to lamellar ichthyosis or congenital ichthyosiform erythroderma. In these cases, the patients reported having symptoms of palmoplantar keratoderma.
3. There is allelic heterogeneity in different genes related to genodermatosis. Molecular biology studies are useful in searching for the aetiology of a possible hereditary condition. Molecular studies help determine an aetiological diagnosis and enable a better understanding of disease pathophysiology, and as a result, aid the identification of more therapeutic options for patients.
INTRODUCTION
Congenital ichthyosis represents keratinisation disorders characterised by abnormal skin scaling across the entire body, leading to a red, denuded, and scaly appearance.1 These disorders include both syndromic and non-syndromic forms, depending on the involvement of additional systems or organs.2 Non-syndromic ichthyoses were traditionally categorised into common ichthyoses (vulgar ichthyosis and non-syndromic X-linked recessive ichthyosis), keratinopathic ichthyoses (epidermolytic ichthyosis, congenital reticular ichthyosiform erythroderma, superficial epidermolytic ichthyosis, and Curth-Macklin ichthyosis), and autosomal recessive congenital ichthyosis (ARCI; lamellar ichthyosis, congenital ichthyosiform erythroderma, harlequin ichthyosis, self-resolving collodion baby, premature baby ichthyosis syndrome, and bathing suit ichthyosis).1,3
Currently, ichthyoses are classified according to the molecular mechanisms that underlie the disease, encompassing alterations in intracellular protein networks (keratins, filaggrin, and cornified envelope), disruption of lipid metabolism (ceramides, cholesterol, lamellar bodies, and dolichol), disturbances in intercellular junctions (tight junctions, desmosomes, and proteases), or impairments in multiple functions. This classification aids in guiding specific treatment options.4
The authors report a familial case involving two sisters, born to non-consanguineous parents, both demonstrating a compound heterozygous variant in the ABCA12 gene, presenting with palmoplantar keratoderma.
CASE DESCRIPTION
Patient 1
A 14-year-old female adolescent who, at birth, presented with collodion membrane and a persistent whitish scaling on palms, soles, skin folds, and joints. Currently, the patient presents with palmoplantar keratoderma accompanied by generalised xerosis and the development of rough hyperpigmented plaques in the axillae, inguinal region, elbows, and knees, along with moderate pain (Figure 1).
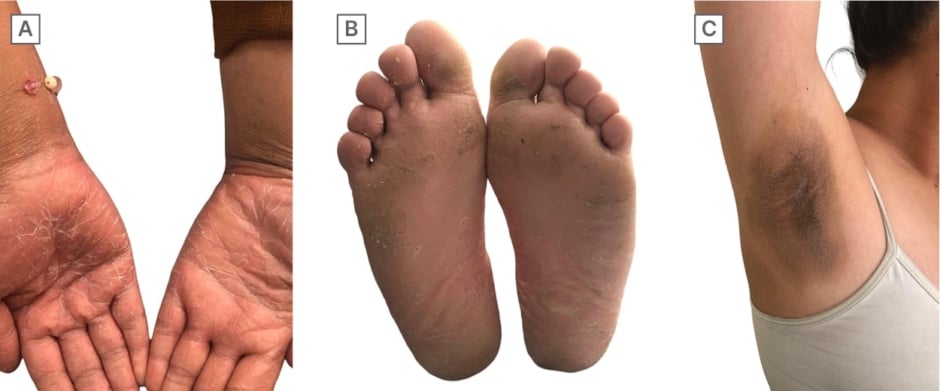
Figure 1: Photos of Patient 1.
A) Palmar keratoderma. B) Plantar keratoderma. C) Rough hyperpigmented plaques in the axillae.
A skin biopsy from the left axilla revealed orthokeratotic hyperkeratosis, papillomatosis, and acanthosis, with areas of hypergranulosis and thick keratohyalin clumps, indicative of a pattern consistent with epidermolytic hyperkeratosis. A biopsy from the left palm demonstrated marked physiological hyperkeratosis settlement in the stratum corneum, accompanied by mild hypergranulosis, consistent with keratoderma. The diagnosis of diffuse isolated palmoplantar keratoderma was established, and management with 20% and 30% urea creams was initiated, maintaining clinical stability.
Patient 2
A 5-year-old female who presented with scaling on palms and soles since birth, currently presenting with palmoplantar keratoderma and generalised xerosis (Figure 2).
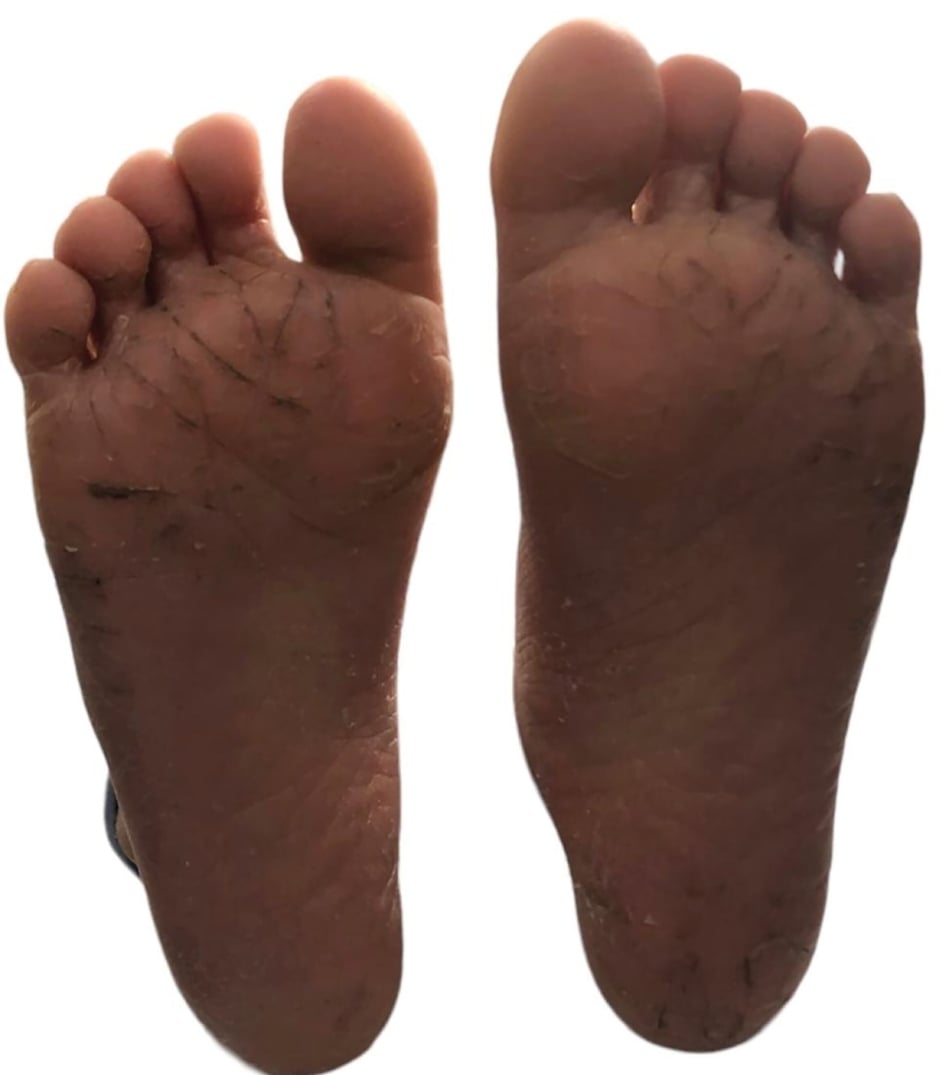
Figure 2: Plantar keratoderma. Photo of Patient 2.
A left palm skin biopsy revealed physiological hyperkeratosis with hypergranulosis, thick keratohyalin clumps, and mild psoriasiform acanthosis, without dermal changes, consistent with keratoderma. The diagnosis of diffuse isolated palmoplantar keratoderma was established, and management was initiated with moisturising creams, 20% and 30% urea creams, along with skin barrier care, resulting in a stable clinical evolution.
Genetic assessment excluded the existence of comparable family histories involving asymptomatic non-consanguineous parents. A Mendelian pattern of incomplete penetrance or germinal mosaicism was considered. A comprehensive trio exome analysis (Patient 1–mother–father) was undertaken, encompassing the study of over 20,000 genes through next-generation sequencing with DNB-SEQG400 (MGI Tech, Shenzhen, China), utilising an MGIEasy Exome Capture V5 Probe Set. Genes associated with established monogenic diseases were scrutinised, maintaining a minimum depth of 20x. A pathogenic nonsense variant in ABCA12 (NM_173076):c.6610C>T (p.Arg2204*), was identified in both the father and the patient, alongside a probably pathogenic intronic variant in ABCA12 (NM_173076):c.6962+2T>C p.?(uncertain protein structure), present in both the mother and the patient.
The molecular findings confirmed the diagnosis of ARCI due to compound heterozygosity in the ABCA12 gene. Each parent carries a pathogenic variant, resulting in a 25% probability of disease recurrence in the patient’s offspring. It was determined that both patients shared the ARCI genotype, but the late onset and clinical manifestations were consistent with the phenotype predominant palmoplantar keratoderma features (Figure 3).
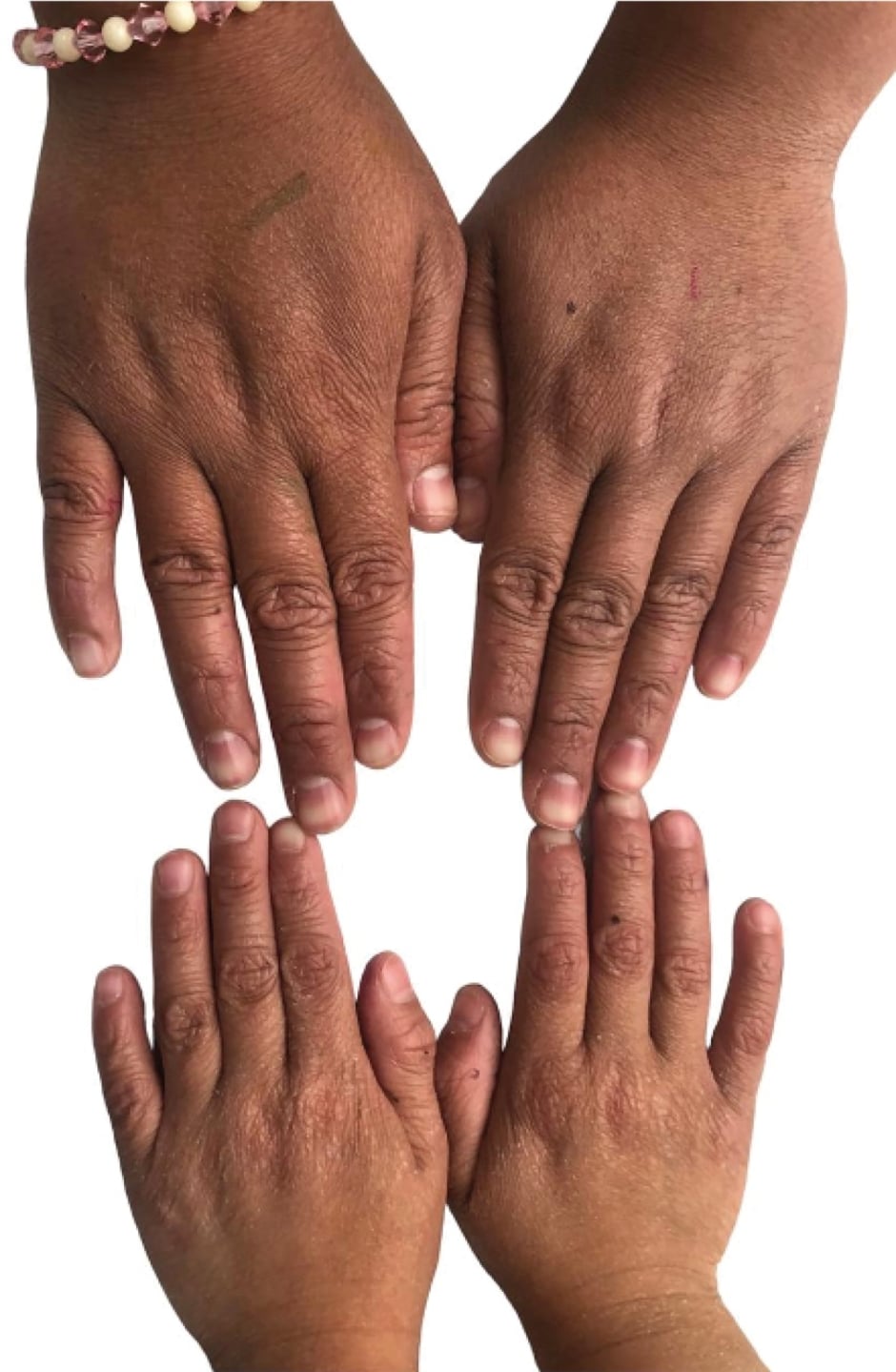
Figure 3: Generalised xeroderma. Photo of Patients 1 and 2.
DISCUSSION
Ichthyoses are distinguished by an aberrant keratinisation process affecting the stratum corneum and stratum granulosum. However, molecular defects associated with barrier impairment and compensatory skin responses vary among ichthyosis types, influencing the signs and symptoms observed in patients.5 The stratum corneum, comprising corneocytes (cellular compartment) surrounded by lipids (intercellular compartment) and interconnected by numerous corneodesmosomes, undergoes proteolytic degradation, a process known as desquamation.6
ARCI constitute a diverse group of congenital ichthyoses devoid of extracutaneous involvement, distinguished by a common molecular mechanism affecting the intracellular protein network, specifically ceramides.4 Collectively, these autosomal recessive disorders have an estimated incidence of 1 in 200,000 live births, resulting in barrier defects that augment transepidermal water loss and trigger compensatory hyperproliferation or hyperkeratinisation.2 The prevalent phenotype within ARCI is lamellar ichthyosis, frequently accompanied by palmoplantar keratoderma, ectropion, and anhidrosis.5 Approximately 60–70% of patients manifest severe symptoms, including a collodion membrane at birth,7 with rare instances of harlequin ichthyosis associated with higher perinatal mortality.5
The compromise of skin barrier function has been associated with pathogenic variants in genes such as ABCA12, ALOX12B, ALOXE3, CERS3, CYP4F22, NIPAL4, PNPLA1, SDR9C7, SULT2B1, TGM1, LIPN, CASP14, and SLC27A4, which play a crucial role in lipid biosynthesis (acylceramides, lipid lamellae, and cornified lipid envelope), intercellular adhesion, and detachment of differentiated cells, among other functions.6
The ABCA12 gene, located on chromosome 2q34, encompasses 53 exons that encode a 2595-amino acid glucosylceramide transporter known as ABCA12. As a member of the ATP-dependent ATP-binding cassette (ABC) transporter superfamily, its primary role is to facilitate the transport of molecules across cell and intracellular membranes.8 The expression of ABCA12 has been demonstrated through RT-PCR methods in placental, testiclular, fetal brain, and skin tissue.8,9 Murine models have revealed that ABCA12 deficiency leads to impaired lipid transfer, disrupting the distribution and transport of glucosylceramides and reducing hydroxyceramide levels. Additionally, it is associated with defects in the transport of certain proteases, such as calpain 5 and cathepsin D, due to alterations in lamellar bodies, contributing to deficient desquamation and hyperkeratosis.10
In humans, ABCA12 is implicated in ARCI,11 specifically associated with harlequin ichthyosis, which represents the most severe and lethal form.12 Frequently observed variants involve large deletions and nonsense variants, resulting in a truncated protein and contributing to the severity of harlequin ichthyosis.13 However, missense variants have also been documented without truncating the protein, leading to milder phenotypes resembling lamellar ichthyosis or congenital ichthyosiform erythroderma with medium-sized scales.5,10
In the author’s case, the identified genetic variants, classified as pathogenic and probably pathogenic following the American College of Medical Genetics and Genomics (ACMG) criteria, consist of nonsense variants featuring premature stop codons and intronic variants, respectively. The former significantly impairs protein function, while the latter results in an uncertain protein. However, when situated in the canonical splicing donor site that disrupts the donor or acceptor splice site, it typically leads to a loss of protein function.14 It likely gives rise to a degraded mRNA transcript or a non-functional protein through mRNA degradation mediated by terminator variants and loss-of-function variants in ABCA12 are known to be pathogenic.8 Because of this, the authors thought both variants were harmful. A computer tool (autoPVS1) showed that the GT-AG splice sites are found in important gene transcripts. If exon skipping or a hidden splice site changes the gene’s reading frame, the mRNA is expected to be broken down by the NMD (nonsense mediated decay) system.15
Hotz A. et al.,15 presented a cohort of 64 patients carrying biallelic variants in ABCA12 with autosomal recessive congenital ichthyosis. The splicing site variation was not reported; however, the nonsense variant (NM_173076):c.6610C>T (p.Arg2204*) was reported in a homozygous patient associated with harlequin ichthyosis. It has been described that loss-of-function variants on both alleles generally result in harlequin ichthyosis, whereas biallelic missense variants mainly lead to lamellar ichthyosis or congenital ichthyosiform erythroderma.15
Despite the protein-level compromise, both variants might suggest a harlequin ichthyosis phenotype. However, the patients in this case exhibited a milder clinical manifestation with a self-limited collodion baby syndrome in the neonatal stage and subsequent development of palmoplantar keratoderma without additional findings. The authors propose that this presentation may be attributed to the compound heterozygous genotype, although experimental studies are recommended to validate this hypothesis.
The management of ARCI is primarily symptomatic, involving the use of topical emollients like glycerol, urea, propylene glycol, sodium chloride, vitamin E acetate, and petroleum jelly, typically formulated in creams.6 In cases of pronounced hyperkeratosis, additional keratolytic agents such as hydroxy acids (lactic and glycolic acids), salicylic acid, N-acetylcysteine, urea (> 5%), and propylene glycol can be introduced. Furthermore, keratinocyte differentiation modulators, including topical retinoids (tretinoin, adapalene, and tazarotene), calcipotriol, and dexpanthenol, may be incorporated into the treatment regimen.10
Presently, investigations are in progress regarding the development of biologic drugs for the treatment of ARCI. Studies involving mice with ABCA12 deficiency, revealing diminished desquamation linked to calpain deficiency, propose that the topical administration of recombinant calpain (Kallikrein-10) appears to reinstate corneodesmosome proteolytic degradation, thereby partially normalising the skin phenotype.16
CONCLUSION
Congenital ichthyosis encompasses a diverse spectrum of pathologies marked by genetic heterogeneity and variable expressivity. Variants in the ABCA12 gene are implicated in autosomal recessive congenital ichthyosis, including congenital ichthyosiform erythroderma, lamellar ichthyosis, and often fatal forms such as harlequin ichthyosis. Despite identifying a genetic variant typically associated with severe clinical phenotypes, the subdued presentation in these patients might be elucidated by their compound heterozygous status.
Presently, the management of ichthyoses remains symptomatic, with the prospect of emerging therapeutics poised to enhance the quality of life for affected individuals. The manifold clinical manifestations and intricate genetic underpinnings underscore the necessity for ongoing research in ichthyosis, aiming to unravel molecular complexities and formulate targeted therapeutic interventions.





