
Abstract
Chronic urticaria is one of the most commonly diagnosed dermatoses. Following diagnosis, correct identification and proper treatment significantly reduces disease activity, thereby improving the patient’s quality of life. However, there is an extensive differential diagnosis for chronic urticaria that, if missed, can lead to life-threatening sequelae. Many of the diseases that masquerade as urticaria are rare and often have a significant delay in diagnosis. This paper aims to fill the gap in the literature by clearly characterising the cutaneous eruptions and atypical findings in many of the most common mimickers of chronic urticaria. Conditions such as erythema marginatum seen in conjunction with hereditary angioedema, urticaria vasculitis, autoinflammatory cryopyrin-associated periodic syndromes, adult-onset Still’s disease and systemic onset juvenile arthritis, Schnitzler syndrome, erythema multiforme, and cutaneous mastocytosis will be discussed.
INTRODUCTION
Urticaria is one of the top ten most commonly diagnosed dermatoses, with up to 20% of the population at risk of developing an acute eruption.1,2 Urticaria is a heterogeneous disorder that, in most cases, is divided into acute or chronic severity of an inducible or spontaneous disease subtype. Urticaria is considered acute (AU) if the lesions last <6 weeks, and potential causes include contact urticaria, food allergies, adverse reactions to drugs, viral infections, and insect bites. If the flare lasts >6 weeks, it is classified as chronic urticaria (CU). CU is characterised by the sudden appearance of wheals or angioedema, and, in up to 40% of cases, both. Hives are evanescent, meaning they usually resolve within 24–48 hours, although sometimes lesions can take longer to dissipate.1-6
CU can be further divided into two major subgroups: spontaneous and inducible. The former occurs without the presence of an exogenous stimulus and can be persistent or intermittent, whereas inducible hives are physical urticarias that can be reproduced by physical stimuli, including cold, sunlight, water, and exercise, or emotional triggers, such as cholinergic or adrenergic urticaria. Hives are considered idiopathic when the history and supportive testing fail to identify an underlying cause.4
There are a number of conditions, presented in Box 1, that can look like CU; therefore, a high index of suspicion must be present to differentiate these rare mimickers from true CU. Early diagnosis of these other conditions increases the chance of successful treatment and unnecessary morbidity and, in some cases, the related potential mortality. Furthermore, proper identification and treatment of CU greatly reduces disease activity, thereby improving patients’ quality of life.
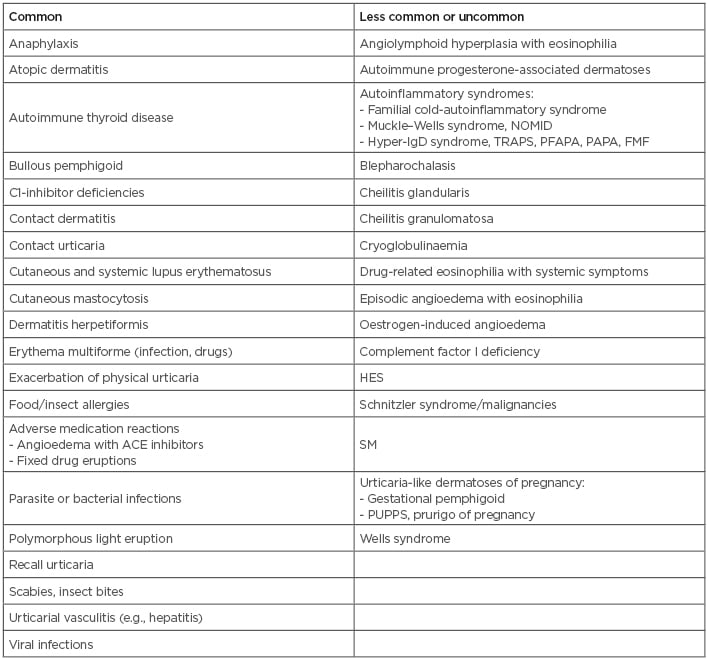
Box 1: Common and less common or uncommon causes of urticaria angioedema and urticaria-like dermatoses.
ACE: angiotensin converting enzyme; FMF: familial Mediterranean fever; HES: hypereosinophilic syndrome; NOMID: neonatal-onset multisystem inflammatory disease; PAPA: pyogenic arthritis, pyoderma gangrenosum, and acne syndrome; PFAPA: periodic fever, aphthous stomatitis, pharyngitis, cervical adenitis; PUPPS: pruritic urticarial papules; SM: systemic mastocytosis; TRAPS: tumour necrosis factor receptor associated periodic syndrome.
Thus, the aim of this paper is to define and discuss the presentation, pathomechanism(s), complications, and treatments of clinical conditions that may be incorrectly diagnosed as CU. These include erythema marginatum (EM) seen in conjunction with hereditary angioedema (HAE); urticarial vasculitis (UV); autoinflammatory cryopyrin-associated periodic syndromes (CAPS); adult-onset Still’s disease (AOSD); systemic-onset juvenile arthritis; Schnitzler syndrome (SchS); erythema multiforme (EMu) cutaneous mastocytosis (CM); and dermatitis artefacta.
ERYTHEMA MARGINATUM
EM is a skin condition commonly seen as a prodromal symptom in patients with a diagnosis of HAE. A recent Danish study by Rasmussen et al.7 found that 56% of HAE patients present with EM as a prodrome.
HAE is a rare autosomal dominant disease that is the result of a dysfunctional C1 esterase inhibitor (C1-INH) protein leading to dysregulation of kallikrein, which is essential for preventing the unimpeded breakdown of high molecular weight kininogen to form bradykinin, a potent vasoactive peptide that binds to bradykinin B2 receptors leading to vascular permeability and recurrent attacks of angioedema.8-16
Several studies have found that physicians struggle to recognise this sentinel rash and other prodromal symptoms of HAE leading to a delay in diagnosis ranging from 8–20 years in >50% of patients.7-9 This can have serious consequences as patients who are misdiagnosed, and therefore not optimally treated, can experience increased mortality related to asphyxiation secondary to laryngeal angioedema.8
A thorough physical exam, as well as patient and family history, provide the subtle clues needed for differentiating CU (Figure 1A) from EM. The EM rash (Figure 1B) is usually asymptomatic, more reticular and serpiginous, less widespread, and typically non-pruritic compared with CU. The most frequently involved sites are the abdomen, axillae, and extremities. Similarly to CU, EM lesions tend to fade within a few hours, but they can persist for as long as 2–3 days; however, unlike CU, which leaves no residual scarring or pigmentation, EM lesions often leave pale or slightly pigmented macules. The EM rash can recur daily over several weeks at different sites of the body, making EM confusing to differentiate from CU. In these patients, a serum C4 level, C1-INH functional and quantitative levels, and a C1Q should be measured to confirm a diagnosis of HAE.7 The exact pathophysiology of EM is not well understood, but it may involve abnormal humoral and cellular immune responses to undefined antigens.7-17
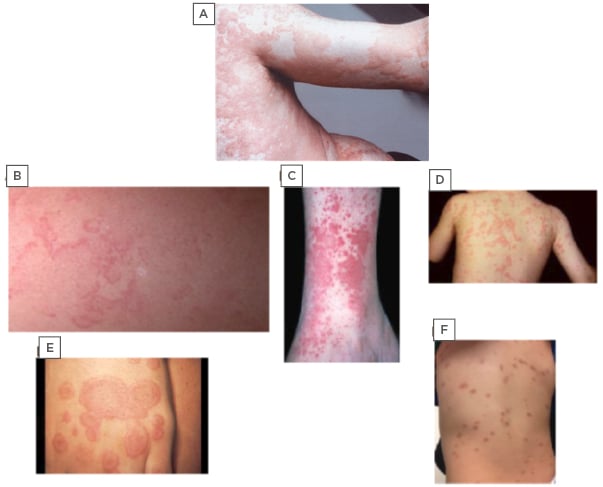
Figure 1: Photographs depicting chronic urticaria and commonly misdiagnosed rarer conditions.
A) Chronic urticaria; B) erythema marginatum; C) urticaria vasculitis; D) familial cold autoinflammatory syndrome; E) erythema multiforme; F) cutaneous mastocytosis.
URTICARIAL VASCULITIS
Up to 15% of refractory urticaria is due to UV, which is estimated to occur in approximately 1% of all treatment refractory CU cases, although the prevalence does vary based on the population studied.17 Urticaria vasculitis includes normocomplementemic UV and hypocomplementemic UV, as well as hypocomplementemic urticarial vasculitis syndrome.
Unlike AU or CU, the rash associated with UV can be described as either painful or burning and lasts longer than 24–48 hours;4,18,19 however, they may have a similar evanescent pattern to typical CU lesions (Figure 1C). The UV lesions differ from CU as they often leave associated bruising or hyperpigmentation and may be associated with a broad range of systemic involvement after resolution of the rash. The extent of extracutaneous manifestations, such as renal and pulmonary involvement, frequently correlates with decreasing levels of complement. Furthermore, up to 50% of patients with hypocomplementemic UV may develop or have coexistent systemic lupus erythematosus or other connective tissue disorders.19,20
Diagnosis of UV requires a consistent clinical presentation and histologic confirmation. Histopathology reveals small-vessel leukocytoclastic vasculitis with fibrinoid, complement, and immunoglobulin deposits perivascularly.18,21 Additional laboratory tests include complete blood count; complete metabolic panel; erythrocyte sedimentation rate (ESR); C reactive protein (CRP); hepatitis profile; urinalysis; and C3, C4, and anti-C1q antibody assays. Depending on the presentation, further tests for connective tissue diseases may be indicated.19,21
Although both CU and UV activate the complement pathway, the latter is a Type 3 hypersensitivity reaction mediated by the deposit of antigen-antibody complexes on the vascular endothelium.21 These antigen–antibody complexes activate the complement pathway, leading to increased anaphylatoxins (C3a, C5a) capable of activating mast cells, resulting in the release of bioactive mediators (i.e., histamine) and urticarial plaques. Just as in CU, the eliciting factor is usually not discovered, but possible aetiologies include medications, infections, autoimmune diseases, and malignancy.18
There are no formal guidelines for the treatment of UV, but antihistamines are often effective to control the pruritus. Other immunosuppressive, anti-inflammatory, or biologic treatments as recommended by the Joint Task Force Urticaria guidelines may be necessary and have been reported to have variable effectiveness.18-21
AUTOINFLAMMATORY SYNDROMES
Autoinflammatory conditions are a group of rare disorders that result from the inappropriate activation of the innate immune system (Table 1).22,23 These conditions include SchS, CAPS, systemic onset juvenile idiopathic arthritis (SoJIA), and AOSD, which all can be easily mistaken for urticaria. However, the rash seen in autoinflammatory syndromes consists of predominantly flat wheals.22 Their exact pathophysiology was not well understood until the recent discovery of IL-1 antagonists, most notably anakinra. The strong response of these conditions to anakinra solidified the notion that these are autoinflammatory conditions mediated by IL-1. A high index of suspicion is needed to recognise the subtle variations of these conditions upon clinical presentation.
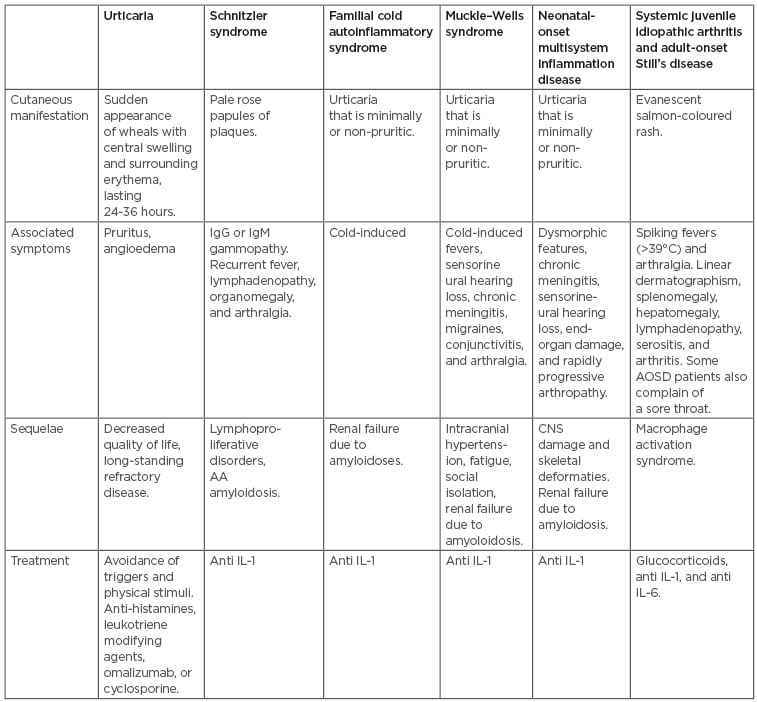
Table 1: Characterisation of urticaria versus autoinflammatory syndromes.
AA: amyloid A; AOSD: adult-onset Still’s disease; CNS: central nervous system.
Schnitzler Syndrome
The clinical picture of SchS can be very difficult to diagnose. Since its first description in 1972, the condition remains relatively underdiagnosed with only 250 known cases and a 5-year delay in diagnosis.24 The mean age of presentation is 51 years, with a slight male predominance.25
SchS can be differentiated from urticaria by the presence of either pale rose macules or papules and plaques that, unlike in urticaria, may range from non to slightly pruritic, completely resolve within 24 hours, and commonly occur in the absence of angioedema. The hallmark feature is the presence of a skin rash with a monoclonal gammopathy, most commonly an IgM gammopathy with very few cases manifesting as an IgG gammopathy.26 Initially, the IgM levels are low and may increase at a rate of roughly 0.5–1.0 g/L/year.25,26 Similarly to the physical urticarias, these skin lesions can be induced by the same triggers, such as cholinergic, cold, and aquagenic stimuli.26
Other common symptoms include fever, arthralgia, and organomegaly. Most patients will develop recurrent fevers either with or separately from the skin flare. Eighty percent of cases exhibit musculoskeletal involvement, with significant bone pain being the predominant complaint and confirmed by radiologic findings in 30–40% of cases. Hepatomegaly and splenomegaly occur in an estimated 30% of SchS patients. Palpable lymph nodes occur in roughly 45% of cases. A definitive diagnosis is made if two obligate criteria (which include a chronic urticarial rash and either an IgG of IgM monoclonal gammopathy) and at least two minor criteria (recurrent fever >39°C, abnormal bone remodelling, neutrophilic dermal infiltrate on skin biopsy, leukocytosis, increased CRP or ESR, hepatomegaly or splenomegaly, lymphadenopathy, arthralgia, or arthritis) in the presence of an IgM gammopathy are met. If the monoclonal component is IgG then three minor criteria are required. Collectively, these requirements for diagnosis are referred to as the Strasbourg Criteria.24-28
Biopsied plaques reveal a perivascular and interstitial neutrophilic inflammation with leukocytoclasia, without vasculitis or dermal oedema, and therefore SchS is classified as a neutrophilic dermatosis. In addition, immunofluorescent studies have reported IgM deposits in 30% of patients.26
In addition to the physical and aesthetic impairments patients suffer, an estimated 20% will develop a lymphoproliferative disorder, such as a monoclonal gammopathy of undetermined significance, and there is a 15% 10-year risk of developing Waldenstrom’s macroglobulinaemia. Other complications include severe inflammatory anaemia and amyloid A amyloidosis.24-28 In the same way that SchS occurs through a distinct mechanism from that of AU or CU, it also requires a different treatment. Recent case studies demonstrate complete resolution of symptoms with treatment using IL-1 inhibitors.21,24-28
Cryopyrin-associated Periodic Syndromes
CAPS encompass three autoinflammatory syndromes: familial cold autoinflammatory syndrome (FCAS), Muckle–Wells syndrome (MWS), and neonatal-onset multisystem inflammatory disease (NOMID). Like other orphan diseases, its true prevalence is hard to estimate and there is a delay in diagnosis. In fact, a study by Stych and Dobrovolny29 found that 44% of FCAS cases carried a different primary diagnosis.
The severity of presentation is variable but includes daily urticaria, fever, and arthralgia. The mildest form is FCAS and the most severe form is NOMID.22,28 CAPS initially presents in infants <6 months. The rash, an atypical urticaria, develops in a circadian pattern as the rash develops throughout the day, and has minimal or no pruritus, hence making it easily confused with UV. However, the dermatologic flare in CAPS, unlike UV, usually resolves within 24 hours without any leftover bruising or hyperpigmentation.18,12-22,28
Each of the three CAPS has its own unique features in addition to the aforementioned presentation. FCAS, similarly to cold-induced urticaria, presents after exposure to cold temperatures (Figure 1D). Both MWS and NOMID have associated sensorineural hearing loss, which is more severe in NOMID patients. Patients with MWS also present with cold-induced fevers and conjunctivitis.22,28-30 NOMID newborns present with dysmorphic features, a myriad of CNS impairments, pseudo-urticaria lesions, end-organ damage, and rapidly progressive arthropathy. The extent of joint involvement can mimic SoJIA in addition to urticaria. The CNS changes include chronic aseptic meningitis, papilloedema, headaches, vomiting, static diplegia, and impaired cognitive function. Brain MRI shows central nervous inflammation, brain atrophy, and enlarged ventricles due to the continuous increase in intracranial pressure.18,20-22,28-30
Just as with NOMID, MWS patients can also present with headaches. In up to 90% of cases, patients complain of headaches due to migraines, chronic meningitis, and intracranial hypertension. In addition, 40–80% of patients suffer from severe fatigue leading to social isolation. Some also experience heaviness of the lower limbs with oedema.22,28-30
A definitive diagnosis of CAPS requires a clinical picture consistent with clinical history and genetic analysis confirming a mutation in the NLRP3 gene. Lab test results usually reveal increased inflammatory markers (CRP and ESR), elevated eosinophils and neutrophils, and low haematocrit. Patients also require frequent urinalysis due to their increased risk of developing systemic amyloid A amyloidosis as a common complication of the disease.20,21,28-30
For treatment of CAPS, IL-1 antagonists have been U.S. Food and Drug Administration (FDA)-approved and are the treatment of choice. Patients treated with these agents are able to live a normal, mostly symptom-free life.18-20,22,28-30
JUVENILE IDIOPATHIC ARTHRITIS AND ADULT ONSET STILL’S DISEASE
SoJIA and AOSD are two similar conditions. SoJIA, a class of juvenile idiopathic arthritis (JIA), occurs before the age of 16 with an age range of presentation between 1 and 16 years. It comprises 10% of JIA cases in northern Europe.31
The triad of a salmon-coloured rash, spiking fevers >39OC, and arthralgia are characteristic of both SoJIA and AOSD. In addition to the classic triad, other clinical manifestations include linear dermatographism, splenomegaly, hepatomegaly, lymphadenopathy, serositis, and arthritis. AOSD patients may initially complain of a sore throat.31-39
Most patients present with an evanescent rash, but some may present with a refractory cutaneous lesion, similar to that seen in CU. This finding can make it difficult to distinguish one from the other. In such cases, the onset of the rash in the evenings concurrently with fever and arthralgia should raise the possibility of either SoJIA or AOSD.
The prognosis of SoJIA and AOSD is complicated by clinical or subclinical macrophage activation syndrome in 10% and 30–40% of patients, respectively. SoJIA and AOSD have a mortality rate of 10–22% and it is therefore imperative that the diagnosis be determined as soon as possible.31-40
With respect to treatment, both AOSD and SoJIA respond well to anti IL-1 or IL-6 therapies and have better outcomes than treating with the traditional disease-modifying antirheumatic drugs. Glucocorticoids are used to treat acute flare-ups.22,24,32-36
Overall, it is often difficult both for the patient and physician to differentiate CU from autoinflammatory syndromes. Table 1 highlights the key characteristics of each of the cutaneous manifestations and associated symptoms seen in the different conditions. Important red flags that may hint at an inflammatory syndrome include inflammation of the anterior eye, uveitis, periorbital oedema, serositis, stomatitis, ulcers, meningeal inflammation leading to headaches, abdominal complaints, arthralgia, myalgia, CNS changes, lymphadenopathy, and fever.
ERYTHEMA MULTIFORME
EMu is a rare, immune-mediated mucocutaneous condition that is most often associated with the herpes simplex virus (HSV) Type 1. Other infectious causes are HSV Type 2, as well as mycoplasma pneumoniae, hepatitis C, and vulvovaginal candidiasis.3,41
Mycoplasma pneumoniae seems to be a more common eliciting factor of EMu in the paediatric population. It may also be drug-induced by non-steroidal anti-inflammatory drugs, sulfonamides, antiepileptics, and antibiotics.41
Unlike CU, EMu presents with targetoid lesions, which appear over 3–5 days and require 1–2 weeks for resolution.15,41 The cutaneous lesions are fixed, as opposed to evanescent. The primary lesions are erythematous and oedematous round papules, surrounded by areas of blanching. These papules enlarge into the characteristic targetoid lesions composed of three concentric colour zones, seen in EMu.41 The outermost ring is erythematous. Within it, there is an oedematous ring that surrounds a red, inflammatory zone, enveloping a dusky centre or blister due to epidermal necrosis (Figure 1E). EMu most commonly presents on the acral extremities in a symmetric distribution with a preference for the extensor surfaces.
These features help differentiate EMu from fatal conditions, such as Stevens–Johnson syndrome and the more widespread toxic epidermal necrolysis. These initially appear with macular EMu-like target lesions on the trunk.41 Therefore, correct and early diagnosis allows for prompt and effective treatment, in addition to halting progression to Stevens–Johnson syndrome and to toxic epidermal necrolysis.
An acute episode of EMu is usually treated with antihistamines and topical corticosteroids to achieve symptomatic relief. Antiviral therapy for HSV infection, administered after the appearance of EMu, does not shorten the course of disease and is therefore not indicated.41 However, in patients affected with recurrent EMu associated with HSV, continuous prophylaxis with antiviral therapy for at least 6 months is indicated. Most patients require 1–2 years of prophylaxis therapy. If nonresponsive, the antiviral dose can either be doubled or substituted with a different antiviral agent.41
Mucosal involvement is seen in 25–60% of EMu cases.3,41 The extent of mucosal involvement also helps to guide treatment. Minimal mucosal involvement is treated with a corticosteroid gel. For patients whose ability to eat or drink is impaired, oral corticosteroids are prescribed. An ophthalmology consultation is required for cases presenting with ocular involvement.41
CUTANEOUS MASTOCYTOSIS
Mastocytosis is a heterogeneous disorder that is characterised by the accumulation of mast cells within different organs, including the skin, bone marrow, gastrointestinal tract, liver, spleen, and lymphatic tissues. Cutaneous mastocytosis is most easily confused with CU.8,15,19,41 It can be distinguished from CU by its characteristic deep brown-orange pigmentation (Figure 1F).15,19,42-44 Often a wheal-and-flare reaction can be elicited by stroking or rubbing the lesion. This is known as a positive Darier sign. It is different from dermatographism in that it affects both clear and lesioned skin.42-45
The exact cause of CM is not known; however, c-Kit mutations (mast stem cell growth factor receptor, also referred to as proto-oncogene c-Kit or tyrosine-protein kinase Kit or CD117) have been identified in biopsies of affected skin.44
Although CM is a relatively benign condition, it can be associated with systemic mastocytosis, especially in adult patients.15,45 If active systemic mastocytosis cases are not treated, it can lead to life-threatening anaphylactoid reactions.
Over 65% of CM cases occur in the paediatric population, in whom this condition is usually transient and resolved by puberty;42,43 however, it can persist and be very disfiguring. In adults who are affected by CM, the treatment depends on disease severity and includes antihistamines, topical corticosteroids, topical calcineurin inhibitors, phototherapy with or without psoralen, and, more recently, certain tyrosine kinase inhibitors (midostaurin).41-45 Water-soluble cromolyn sodium cream and aqueous-based cromolyn sodium skin lotion have been reported to alleviate pruritus and flares with variable success.
DERMATITIS ARTEFACTA
Although ultimately a diagnosis of exclusion, dermatitis artefacta (DA) should also be considered in the differential diagnosis of urticaria, especially in cases where the past medical history is inconsistent with CU. DA is a factitious disorder, which is also part of a spectrum of psychocutaneous disorders. While urticaria can be elicited by psychosocial stressors as well, DA is self-inflicted and is usually differentiated from other dermatologic conditions via a strong anamnesis and patient–physician rapport. The lesions are non-healing, located in areas that can be easily accessed by the patients’ dominant hand, and surrounded by intact skin. Upon presentation, the patient may seem indifferent to the lesions, while the family is concerned. Moreover, the patient may have other psychiatric illnesses, such as eating disorders and personality disorders.41-43 Patients with DA are at increased risk of harming themselves further and therefore it should not be taken lightly.
CONCLUSION
The conditions discussed in this paper have been historically confused for CU. Although these mimickers are relatively rare, patients’ prognoses can be profoundly affected if they are misdiagnosed. Maintaining a high index of suspicion, especially in cases of refractory urticaria, allows for better treatment and, in many cases, remission of symptoms.





