
Abstract
Hypertrophic scar (HTS) is a dermal form of fibroproliferative disease that develops after deep burns, skin injuries, and surgical wounds. HTS is defined as a visible, elevated scar that does not spread into surrounding tissues and often regresses. HTS often leads to physical and psychological issues for patients. Cellular and molecular agents, such as chemokines, bone marrow derived stem cells, TGF-β, fibroblast heterogeneity, toll-like receptor 4, and small leucine-rich proteoglycans, have been known to play an important role in wound healing and abnormal scar formation. Targeting these agents could have therapeutic potential for the treatment of pathological scars. HTS often causes contracture deformities, aesthetic disfigurement, and prolonged periods of hospitalisation and rehabilitation, leading to psychological complications. Numerous treatments have been described for HTS, but the optimal treatment has not yet been established. Chemokines are implicated in all stages of wound healing, but hyperactivity of these agents leads to excessive scarring. A better understanding of the mechanisms of chemokine action, such as the role of the signalling agents in wound healing and abnormal scar formation, will help to establish more effective therapeutic strategies for HTS.
INTRODUCTION
Hypertrophic scar (HTS) is a dermal form of fibroproliferative disease that develops after deep burns, skin injuries, and surgical wounds. HTS is defined as a visible, elevated scar that does not spread into surrounding tissues. It often causes contracture deformities, aesthetic disfigurement, and a prolonged period of hospitalisation and rehabilitation, which can lead to psychological complications.1,2 Numerous treatments have been described for HTS, but the optimal treatment has not yet been established.
Cellular and molecular factors, such as chemokines, bone marrow derived stem cells (BMSC), TGF-β, fibroblast heterogeneity, toll-like receptor 4 (TLR4), and small leucine-rich proteoglycans (SLRP), are known to play important roles in wound healing and abnormal scar formation; therefore, targeting these agents has therapeutic potential for pathological scars. Chemokines in particular are implicated in all stages of wound healing and hyperactivity leads to excessive scarring. Having a better understanding of the mechanisms of chemokine pathways in wound healing and abnormal scar formation will help to establish more effective therapeutic strategies for HTS.
This review will summarise cellular and molecular mechanisms in wound healing and HTS formation and describe the possibility of targeting the chemokine pathways as a therapeutic approach for HTS.
WOUND HEALING AND HYPERTROPHIC SCAR FORMATION
Normal wound healing consists of four overlapping phases: haemostasis, inflammation, proliferation, and remodelling. Wound healing requires complex interactions among keratinocytes, fibroblasts, the extracellular matrix (ECM), and stem cell populations within the basal epidermis, appendages, and underlying mesenchyme.3 These interactions are coordinated and regulated by a variety of molecules, including growth factors, cytokines, and chemokines.
The inflammatory phase presents as erythema, swelling, fever, and pain around wound sites, which usually lasts 2–4 days after injury. During inflammation, leukocytes leave adjacent blood vessels and migrate to the wound sites, where the cells facilitate the removal of both micro-organisms and cellular debris and secrete a plethora of cytokines that are critical for proper healing. This phase of healing is mediated via growth factors, cytokines, and chemokines, which induce the recruitment of different cell types to the wound sites to promote wound healing.4
The proliferation phase involves fibroplasia, angiogenesis, and the formation of the ECM and the granulation tissue. Fibroplasia is the process whereby fibroblasts proliferate in the wound sites and excrete glycoprotein and collagen, which combine to form the ECM. Angiogenesis is essential to replace damaged capillaries and restore the supply of oxygen, blood constituents, and nutrients to wounded tissue; this process drives the return to normoxia and promotes fibroplasia.5 Angiogenesis simultaneously takes place, which involves the migration of endothelial cells to the wounded area. During the formation of granulation tissue, fibroblasts are activated and differentiate into myofibroblasts, which are highly contractile cells that produce an abundance of ECM proteins. Myofibroblasts contribute to wound contraction, which is an essential process in wound healing.
The remodelling phase begins 2–3 weeks after injury and can last for a year or more. During the remodelling phase, granulation tissue is converted into a mature scar. Type III collagen, the primary component of the granulation tissue, is replaced by type I collagen, which is the primary structural component of the dermis.6 Disorganised collagen fibres are rearranged, cross-linked, and aligned along tension lines. Hypercellularity in remodelling wounds is reduced by apoptosis of residual inflammatory cells, vascular cells, and myofibroblasts.7
Pathological wound healing, mainly caused by chronic inflammation or infection, can lead to fibroproliferative disorders, such as HTS and keloids.8 Increased vascular permeability, which is the result of dilatation of the gaps between the endothelial cells of the vascular wall, allows inflammatory cells to migrate into the wound site. This endothelial function is intensified by genetic mutation, systemic factors, and local factors, such as skin tension. Endothelial dysfunction increases the number of inflammatory cells and the levels of many soluble inflammatory factors that can enter the wound site, thereby increasing local inflammation.9 In some excess healing conditions, the myofibroblasts fail to undergo apoptosis, leading to their prolonged presence in the wound and production of excessive ECM, resulting in fibrosis and abnormal scar formation.10
EPIDEMIOLOGY
It is estimated that HTS develops in up to 67% of burn patients with major burns, as defined by the American Burn Association (ABA). The rate is higher in non-white patients and the lowest incidence of HTS is found in patients with albinism.11 Children have a higher rate of HTS formation than adults.12
AETIOLOGY
An analysis of surgical burn treatment showed that burn depth, type of skin graft; delayed wound healing caused by infection or prolonged inflammation, and wound tension are risk factors for HTS.13,14 BMI, non-Caucasian ethnicity, and scar related factors are positively associated with HTS formation. On the other hand, antihypertensive therapeutics and factors influencing erythropoiesis were negatively associated with HTS formation in patients who underwent elective cardiothoracic surgery.15
CLINICAL SYMPTOMS
Morphologically, HTS presents as a red, hyperaemic, elevated, firm scar and pruritic lesions that have an abnormal texture, which have lost the pliable and elastic attributes of healthy skin. HTS often causes contracture deformities, aesthetic disfigurement, a prolonged period of hospitalisation and rehabilitation, and psychological issues2,8 (Figure 1).
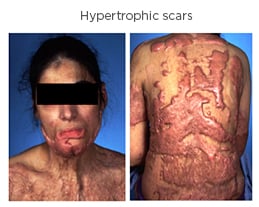
Figure 1: Hypertrophic scars in a 28-year-old woman with after a burn injury.
HISTOLOGY
The main histological features of HTS are a thicker epidermis and dermis; a lack of rete ridges in the epidermis; increased cell proliferation; excess blood vessel formation; and an atypical ECM, featuring increased collagen deposition and thin disorganised collagen fibres in the dermis (Figure 2).16 Collagen bundles contain a greater amount of type III collagen than normal skin and are orientated parallel to the epidermal surface. Fibroblasts and myofibroblasts play essential roles in fibrotic diseases due to the ability of these cells to generate excessive collagen in abnormal wounds; additionally, fibroblasts are the primary contributors of collagen deposition at the wound site. Ultrastructural nodules and large numbers of myofibroblasts expressing alpha smooth muscle actin are seen in the histology of HTS.16
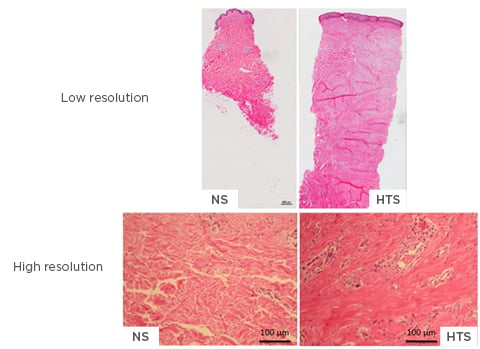
Figure 2: High and low-resolution imaging of normal skin and hypertrophic scarring.
The main histological features of hypertrophic scarring are a thicker epidermis and dermis, lack of rete ridges in the epidermis, increased cell proliferation, excess blood vessel formation, and atypical ECM, such as collagen deposition, and thin disorganised collagen fibres in the dermis. Low resolution: 10x magnification; high resolution: 40x magnification.
ECM: extracellular matrix; HTS: hypertrophic scarring; NS: normal skin.
CURRENT TREATMENTS
Numerous treatments have been described for HTS. Among the therapeutic options are surgical excision, pressure therapy, intralesional interferon, corticosteroids, bleomycin, 5-fluorouracil injections, combination of cryotherapy with intralesional triamcinolone acetonide, silicone gel sheeting, onion extract and heparin gel administration, and laser therapy.17 Currently, the most widely accepted therapeutic modalities are surgery plus adjuvants, such as intralesional steroids, silicone gel sheeting, pressure, and radiotherapy. Despite these treatments, HTS remains difficult to manage and there is no universally accepted treatment regimen.17
BONE MARROW DERIVED STEM CELLS IN HYPERTROPHIC SCARRING
BMSC, such as mesenchymal stem cells, endothelial progenitor cells, and fibrocytes, are involved in the wound healing processes, contributing to skin cells or releasing regulatory cytokines.18 By co-culturing layered fibroblasts with bone marrow derived-mesenchymal stem cells (BM-MSC), the BM-MSC enhanced the fibrotic behaviour of deep dermal fibroblasts.19 Otherwise, BM-MSC conditioned medium decreased expression of profibrotic genes, including connective tissue growth factor, plasminogen activator inhibitor-1, TGF-β1, and TGF-β2 in HTS fibroblasts. In contrast, the expression of antifibrotic genes, such as TGF-β3 and DCN, were substantially enhanced.20 BM-MSC enhance wound healing and prevent HTS formation via inflammatory regulation, differentiation, and angiogenesis and/or release of paracrine factors.19 These studies suggest the involvement of BM-MSC in the pathogenesis of hypertrophic scarring and suggest their therapeutic potential for HTS.
TGF-β IN HYPERTROPHIC SCARRING
TGF-β has recently been shown to be a key factor in modulating contraction in normal skin fibroblasts.6 There are three mammalian isoforms of the growth factor: TGF-β1, TGF-β2, and TGF-β3; each of the isoforms have different effects on the same cells. TGF-β strongly promotes the chemotaxis of fibroblasts to the site of inflammation to produce ECM proteins. Myofibroblasts are an important source of ECM in the development of fibrosis. The transition from fibroblasts to myofibroblasts is influenced by TGF-β1, which is a profibrotic mediator and regulator of the wound healing processes at cellular level, including the regulation of proliferation, differentiation, migration, cell survival, and angiogenesis.6 Dysregulation of TGF-β production or activity can cause abnormal fibrosis. Fetal wounds, which heal without scarring, contain lower levels of TGF-β than adult wounds. HTS tissue expressed five times as much TGF-β1 mRNA as normal skin tissue. In vitro cultured fibroblasts derived from the HTS also expressed TGF-β1 mRNA at a level significantly higher than that of the normal fibroblasts.21 In fetuses that heal without a scar, the ratio of TGF-β3 to the TGF-β1 and TGF-β2 isoforms is high, while in wound healing, the ratio of TGF-β3 to TGF-β1 and TGF-β2 is low, with expression of TGF-β3 emerging at a later stage of the healing process.22 TGF-β3 is a potential mediator of scar reduction and improvement, with the isoform having been used in a surgical model of cleft lip in mouse and sheep fetuses.23 Human recombinant TGF-β3 (avotermin) was shown to promote the regeneration of normal skin and improve scar appearance in Phase I and II trials.24 Both in vitro and in vivo models have demonstrated that avotermin affects a number of processes, including the inflammatory response, cell migration, and protein deposition in the ECM, which collectively result in scar improvement. However, avotermin failed in Phase III clinical trials.24
Bone morphogenetic proteins and activins belong to the TGF-β superfamily and are involved in almost every phase of wound healing. These molecules promote epidermal hyperthickening and re-epithelialisation, keratinocyte proliferation, granulation tissue formation, epithelial to mesenchymal transition, and ECM production. Activins are upregulated after skin injury and production of these proteins is increased in HTS.25
FIBROBLAST HETEROGENITY IN HYPERTROPHIC IN HYPERTROPHIC SCARRING
Fibroblasts synthesise the ECM of connective tissue and play an essential role in maintaining the structural integrity of most tissues. Fibroblasts isolated from different tissues in the body belong to morphologically and functionally heterogeneous subpopulations. Furthermore, these differences seem to be dictated by the local biological and physical microenvironment the fibroblasts reside in. In vitro, deep dermal fibroblasts were found to contain higher levels of mRNA of type 1 collagen, alpha smooth muscle actin, TGF-β, stromal cell-derived factor 1, and tissue inhibitor of metalloproteinase 1, an inhibitor of collagenase.19 Furthermore, deep dermal fibroblasts express a higher number of TGF-β type II receptors; have lower collagenase activity; and produce more fibrotic factors, including TGF-β1, connective tissue growth factor, osteopontin, biglycan, and versican, and less antifibrotic SLRP.19 Fibroblast heterogeneity is necessary for scarless wound healing, complete restoration of native tissue architecture in the fetus and oral mucosa, and excessive scar formation, such as that seen in HTS and keloids.3
TOLL-LIKE RECEPTORS IN HYPERTROPHIC SCARRING
It is understood that prolonged inflammation contributes to HTS formation. Fibroblasts regulate immune and inflammatory responses through TLR4, which is activated by lipopolysaccharide through adaptor molecules, leading to NFκB and mitogen-activated protein kinase activation, cytokine gene transcription, and co-stimulatory molecule expression, which results in inflammation. Dermal fibroblasts from HTS express TLR, and the associated intracellular signalling molecules express at a higher level than normal. As a result, lipopolysaccharide-stimulated fibroblasts express proinflammatory cytokines, including prostaglandin E2, IL-6, IL-8, and monocyte chemoattractant protein-1, which cause persistent inflammation in burn-injured tissue, thus contributing to HTS development.26
EXTRACELLULAR MATRIX COMPONENTS IN HYPERTROPHIC SCARRING
The ECM is a non-cellular, three-dimensional, macromolecular network composed of collagens, elastin, fibronectin, laminins, proteoglycans/glycosaminoglycans, and several other glycoproteins. The ECM has many functions, such as storage and delivery of growth factors and cytokines, tissue repair, and various other physiological functions. Abnormal ECM reconstruction leads to HTS.27 The breakdown of the ECM is an essential component of wound healing and scar formation. Matrix metalloproteinases (MMP), classified by their substrate specificity, catalyse the hydrolysis of major ECM molecules. Furthermore, MMP affect many biological functions by regulating growth factors and their receptors, cytokines and chemokines, cell surface proteoglycans, and other enzymes. MMP are secreted by keratinocytes, fibroblasts, and inflammatory cells during the wound healing process. Mice deficient in MMP, including MMP-3, MMP-8, and MMP-14, showed delayed or impaired wound healing. In HTS, MMP-1, MMP-2, and MMP-9 are decreased and tissue inhibitors of MMP-1 are increased.27
SLRP, such as decorin, biglycan, fibromodulin, and lumican, are ECM molecules that bind to type I collagen to regulate collagen fibrillogenesis and inhibit TGF-β1 fibrogenic activity.6,28 Under healthy conditions, higher levels of decorin and fibromodulin have been found in the superficial layers of the skin compared with levels recorded in the deeper dermal layers. There is an increased expression of biglycan, fibromodulin, and lumican in the basement membrane and around basal epithelial cells. These proteoglycans are absent or weakly expressed in HTS compared to normal skin. Decorin knock-out mice showed a significant increase in the number of fibroblasts in their periodontal ligaments compared with wild-type mice. Decorin deficiency leads to impaired angiogenesis in injured mouse corneas. These findings suggest that down-regulation of SLRP after deep injuries to the skin play an important role in the development of HTS.28
THE ROLES OF THE CHEMOKINE PATHWAY IN HYPERTROPHIC SCARRING
In recent years, many scientific research papers have described the roles of chemokines in the wound healing processes but also various other biological processes and diseases, such as HIV-1, cardiovascular diseases, cancer, atherosclerosis, inflammatory bowel diseases, and rheumatoid arthritis.29-31 Chemokines are a family of small chemotactic cytokines, 8–10 KDa in size, which have been classified into four main subfamilies: C (XCL), CC (CCL), CXC (CXCL), and CX3C (CX3CL) chemokines. The chemokines play critical roles in many basic biological processes, including leukocyte trafficking and homing, tumourigenesis and metastasis, inflammation, autoimmune response, and viral infection.29-31
The normal wound healing processes are regulated by numerous bloodborne cells; the recruitment of these cells is tightly regulated by chemokines. The chemokines are important modulators of each phase in human wound healing.4,29,32 A lack of regulation of the chemokine network can result in chronic inflammation, dysregulation of vessel development, and establishment of a chronic environment that leads to impaired healing, such as HTS, keloids, scleroderma, psoriasis, and various types of fibrosis and chronic wounds.10 HTS is hypercellular due to increased numbers of fibroblasts and the recruitment of peripheral non-haematopoietic cells. The recruitment of bloodborne cells suggests that chemokines may have roles during HTS development.32
After injury, the haemostasis phase starts immediately, and platelets and plasma fibronectin are released along with prothrombin to form a clot. During this phase, CXCL4 plays an important role. CXCL4 has weak chemotactic potency, but it is strongly involved in angiogenesis, haematopoiesis suppression, inhibition of collagenase activity, and accumulation of deleterious lipoproteins at sites of vascular injury. CXCL4 inhibits local antithrombin III activity and promotes coagulation by neutralisation of heparin-like molecules on the endothelial surface of blood vessels.4,29,32
The inflammatory phase is mediated via growth factors, cytokines, and chemokines, which induce the recruitment of different cell types to the wound sites to promote healing.10 Recruitment of proinflammatory cells, such as neutrophils and macrophages derived from monocytes, to the injured sites is necessary to remove debris and kill bacteria through phagocytosis and free radical production. These leukocytes are mainly directed by CXC chemokines, such as CXCL1, CXCL5, and CXCL8, that are stored in blood platelets and are immediately released upon cell activation. CXCL8 was the first chemokine identified as a mediator of the directional migration of leukocytes to sites of inflammation and injury. CXCL8 also activates endothelial permeability through both modulation of adherens junction endothelial cell adhesion and subsequent cell contractions that promote leukocyte diapedesis from the circulation.30 In humans, CXCL8 is one of the main chemoattractants for neutrophils through both the CXCR1 and CXCR2 receptors and stimulates neutrophil activation through CXCR1.31,33
Neutrophil chemotaxis is followed by monocyte chemotaxis to the wound sites, primarily via the CCL2 protein: monocyte chemoattractant protein-1. Monocyte chemoattractant protein-1 is released by neutrophils during the early stages of wound healing, by the monocytes themselves, and by keratinocytes during the later stages of wound healing.34 In the wound sites, monocytes differentiate into macrophages, which phagocytose apoptotic neutrophils and other dead cells, and secrete cytokines, growth factors, and chemokines that promote the latter stages of inflammation and wound repair. Macrophages also stimulate angiogenesis and fibroplasia and ECM production.35
Re-epithelialisation occurs during inflammation and the formation of the granulation tissue. The process involves keratinocyte migration and proliferation to cover the wound area. CXCL11 and its receptor CXCR3 are expressed by basal keratinocytes during re-epithelialisation and promote re-epithelialisation as a mediator of epidermal-dermal communication during wound repair.4,36 CXCR3 knock-out mice showed delayed re-epithelialisation and basement membrane regeneration after wounding.36
CCL2 binds CCR2 presented on endothelial cells and has a role in angiogenesis. CCL2 is produced by a number of cell types, including fibroblasts and endothelial, epithelial, smooth muscle, mesangial, astrocytic, monocytic, and microglial cells. This pathway mediates neovascularisation by affecting membrane type 1-MMP.37 This pathway also regulates migration and infiltration of monocytes into the foci of active inflammation in the wound healing process,38 and has a pathogenic role in the induction of multiple types of fibrosis, such as HTS, keloids, scleroderma, liver cirrhosis, atherosclerosis, pulmonary fibrosis, renal fibrosis, and colon fibrosis. CCL2 knock-out mice form minimal scars, indicating that the inflammatory chemokine pathways are a major mechanism by which focal adhesion kinase mechanotransduction induces fibrosis.39
CCL5 (regulated on activation, normal T cell expressed and secreted) interaction with CCR5 also has a role in angiogenesis. CCL5 increases vascular endothelial growth factor expression and production of human osteosarcoma cells.40 CCR5 is expressed on the surface of various immune cells, including macrophages, monocytes, microglia, dendritic cells, and active memory CD4+ T cells. Endothelial progenitor cell recruitment to the wound site is regulated by CCR5.41 In mice, CCR5 gene expression and its ligands are upregulated at the wound site and the CCR5 protein is exclusively detected in macrophages and endothelial cells. Deficiency of CCR5 reduced endothelial progenitor cell recruitment and attenuated neovascularisation and collagen production in skin wounds. This resulted in delayed wound healing.41
CXCL12 (stromal cell-derived factor-1) is expressed in the epidermis, dermis, hair follicles, blood vessels, and sweat glands. CXCL12 specifically binds to CXCR4 receptors on endothelial cells and induces endothelial cell chemotaxis, which is important for vascularisation. CXCL12 is involved in the homing of BMSC to the wound site during skeletal, myocardial, vascular, lung, and skin wound repair via CXCR4.29
The CXCL12–CXCR4 pathway is involved in the development of HTS by promoting the migration of activated CD14+ CXCR4+ cells from the bloodstream to wound sites, where these differentiate into fibrocytes and myofibroblasts, contributing to the development of HTS.42 Local dermal fibroblasts are activated by inflammatory signals after injury and produce CXCL12, which attracts CXCR4-expressing monocytes to the wound site from the systemic circulation. The CXCL12–CXCR4 pathway is upregulated during wound healing in burn patients and in HTS tissue compared with normal skin.42 Blocking the CXCL12–CXCR4 pathway by CTCE-9908 (a CXCR4 antagonist) minimised HTS formation in mouse model. CTCE-9908 reduced the accumulation of macrophages and myofibroblasts, enhanced the remodelling of collagen fibres, and downregulated the gene and protein expression of fibrotic growth factors in the skin grafts43 (Figure 3). The endogenous stem cell mobilisation produced by AMD3100 (CXCR4 antagonist) and low-dose tacrolimus reduced the time to complete healing of full-thickness wounds by 25% in mice and resulted in fewer scars.44
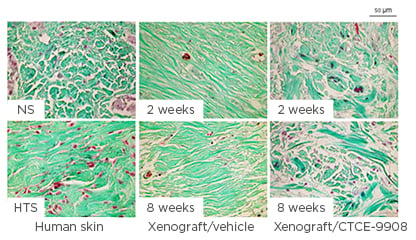
Figure 3: Collagen fibre morphology in the dermis of xenografts.
Representative Masson’s trichrome stain images for collagen fibres in the dermis of xenografts. Masson’s trichrome stain was performed in paraffin-embedded human normal skin, human hypertrophic scarring, and xenograft biopsies collected from mice treated with CTCE-9908 or vehicle at 2 and 8 weeks after grafting, which stains keratin red, collagen green, cell cytoplasm pink, and cell nuclei dark brown.
HTS: hypertrophic scarring; NS: normal skin.
In the remodelling phase, reduction in hypercellularity is controlled by the interaction of CXCL10 and CXCL11, which are respectively produced by maturing endothelium and keratinocytes, with the CXCR3 receptor.4,37 Stimulation of CXCR3 signalling converts fibroblasts from a migratory to a contractile state after an increase of mature dermal collagen fibres, increases keratinocyte migration by activation of m-calpain, and inhibits endothelial cell migration and proliferation.4 In an in vivo study,45 full-thickness excisional wounds in CXCR3 knock-out mice healed with dermal hypercellularity and presented with immature matrix components. They also presented with poor remodelling and reorganisation of collagen, which resulted in a healed dermis that lacked tensile strength. It is hypothesised that hypertrophic scarring results from the fibroplasia and its overproduction of ECM, secondary to abnormalities in epidermal-dermal communication. Signalling through the CXCR3 receptor plays a major role in wound maturation; the absence of this signalling system results in an immature and hypercellular dermis, and HTS characterised by ongoing wound regeneration, cellular proliferation, and scars in which immature matrix components are undergoing increased turnover resulting in a chronic inflammatory process.45
POTENTIAL THERAPEUTIC STRATEGY FOR HYPERTROPHIC SCARRING IN THE FUTURE: TARGETING CHEMOKINE PATHWAYS
Chemokines are essential for wound healing and scar formation by coagulation, leukocyte infiltration, re-epithelialisation, angiogenesis, stem cell recruitment, collagen reorganisation, and lymphocyte-mediated immune response. After dermal injury, bloodborne cells are recruited to the wound sites from systemic circulation by chemokines, such as CXCL12, which is released from dermal fibroblasts. These cells are considered as progenitor cells of macrophages and fibrocytes and contribute to not only wound healing but also hypertrophic scarring. The regulation of these processes by inhibition of chemokine pathways, including the CXCL12–CXCR4, CCL2–CCR2, and CCL5–CCR5 mechanisms, has the potential to reduce hypertrophic scarring. These pathways are a potential therapeutic target for HTS.





