Meeting Summary
In this interactive symposium and question and answer session at the European Society of Cardiology (ESC) Congress 2020, co-chair Dr Howard gave an overview of pulmonary hypertension (PH) and pulmonary arterial hypertension (PAH). He highlighted the need for early intervention while right ventricular (RV) function is preserved. Dr Escribano addressed the challenges faced in the diagnosis of the different forms of PH, particularly the difficulties in distinguishing patients with PAH from those with PH due to left heart disease (LHD). She detailed a three-step pragmatic approach for their differential diagnosis: 1) identification of a specific PH-LHD phenotype; 2) definition of a pretest probability of PH-LHD; and 3) haemodynamic assessment. Prof Rosenkranz highlighted the implication of right heart remodelling on long-term outcomes, as well as the treatment strategies to improve RV function; regular RV imaging (echocardiography, cardiac MRI [cMRI]) and haemodynamic assessments are recommended. Further, early diagnosis of PAH and prompt initiation of treatment to maintain or improve RV function are important for patient outcomes.
Prof Galiè presented a risk management-based approach to treatment decisions and outlined the critical importance of improving long-term outcomes in PAH through timely treatment choices. He highlighted key messages from several studies that have shown beneficial effects of PAH-specific treatments (e.g., double combination therapy with macitentan and a phosphodiesterase-5 inhibitor [PDE-5i] or triple combination therapy with selexipag, a PDE-5i, and an endothelin receptor antagonist [ERA]) on morbidity/mortality events in PAH. He emphasised that choosing the right treatments is key to achieving the best long-term outcomes in PAH. In her closing remarks, co-chair Dr Lang underlined that current treatment regimens are based on appropriate risk assessments that need to be repeated every 3–6 months, and that nowadays, we have learned to combine medications upfront and that “more is better.”
Introduction
Doctor Luke Howard
PH encompasses a heterogeneous group of conditions, including PAH (Group 1), PH due to LHD (Group 2), PH due to lung diseases and/or hypoxia (Group 3), PH due to pulmonary artery obstructions (Group 4), and unclear/multifactorial mechanisms (Group 5).1,2 Patients, particularly those with LHD, can present with multiple comorbidities,3 which complicate assessment and treatment. Patients can also have characteristics of more than one group, which can be challenging. For example, there may be a continuum from typical idiopathic PAH (IPAH) (Group 1), through atypical IPAH, to PH due to heart failure with preserved ejection fraction (HFpEF) (Group 2), on which patients have a declining precapillary component, an increasing risk factor profile, and may have declining efficacy and increasing side effects of targeted PAH therapy.4
Intervention early in the course of PH, while a patient has preserved RV function, is likely beneficial as a patient’s condition can decline precipitously in later stages.5 Regular assessment of RV function can be used to assess treatment response and identify patients whose condition is deteriorating.6 Regular risk assessment allows for early intervention and prompt treatment intensification, resulting in improved functional capacity and prognosis, and a decreased risk of progressive remodelling and right heart failure.7
Making the Correct Diagnosis: Differentiating Pulmonary Arterial Hypertension from Pulmonary Hypertension Due to Left Heart Disease
Doctor Pilar Escribano
Globally, LHD is the most common cause of PH.8 Among patients with LHD, PH is related to the severity of the underlying condition and impacts on symptoms, exercise capacity, treatment decision-making, and prognosis. LHD can cause PH when left ventricular (LV) dysfunction increases LV filling pressures, resulting in passive backward transmission to the pulmonary circulation and increased pulmonary pressure.9 Ultimately, this can impact on the right heart, potentially resulting in RV failure, which is associated with a poor prognosis.9
PH-LHD is classified as postcapillary PH (mean pulmonary arterial pressure >20 mmHg; pulmonary arterial wedge pressure [PAWP] >15 mmHg), indicating backward transmission of increased left-sided pressure. Two types of postcapillary PH have been defined: isolated postcapillary PH (pulmonary vascular resistance [PVR] <3 Wood Units [WU]) and combined pre- and postcapillary PH (PVR ≥3 WU).1 The former is easily distinguished from PAH, but the latter has much more overlap.
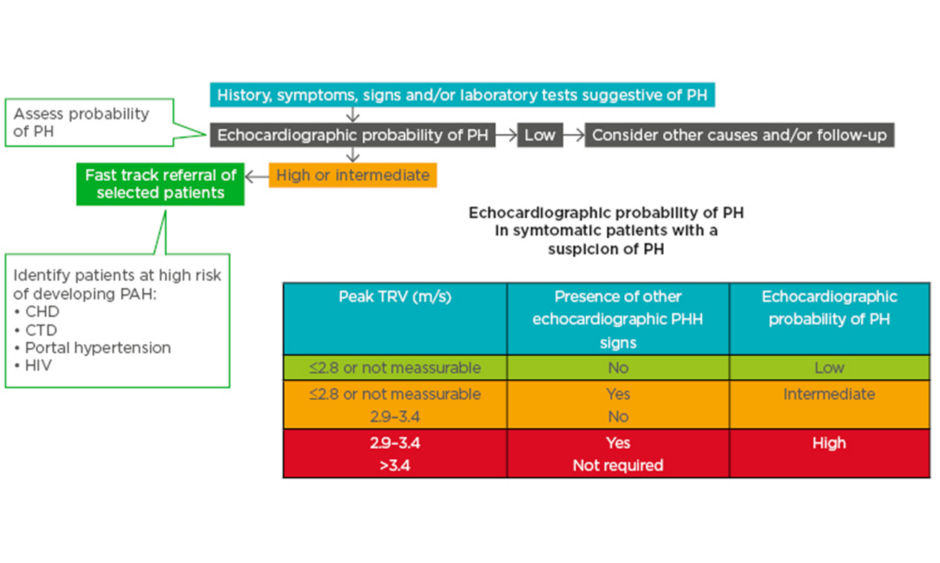
Echocardiography can be used to ascertain the probability of PH in patients with suspected PH (Figure 1).10 Patients with high/intermediate probability who are part of a risk group (congenital heart disease, connective tissue disease, portal hypertension, and HIV infection) are recommended for fast track referral (Figure 1).10
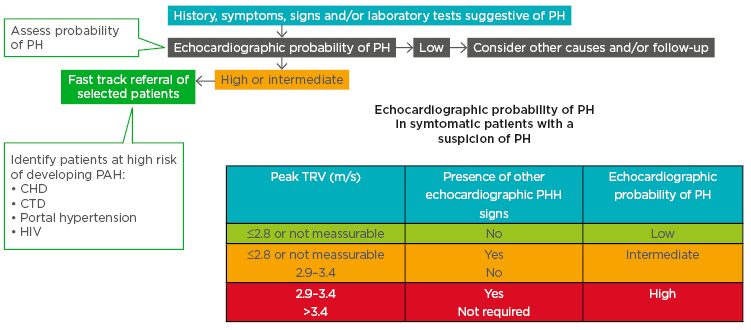
Figure 1: A fast-track referral is recommended for patients with a high pretest probability for pulmonary arterial hypertension.
CHD: congenital heart disease; CTD: connective tissue disease; PAH: pulmonary arterial hypertension; PH: pulmonary hypertension; TRV: tricuspid regurgitation velocity.
This material has not been reviewed prior to release; therefore the European Respiratory Society may not be responsible for any errors, omissions or inaccuracies, or for any consequences arising there from, in the content. Reproduced with permission of the © ESC &ERS 2020. European Respiratory Journal Oct 2015, 46 (4) 903-975; DOI: 10.1183/13993003.01032-2015.
Although various echocardiographic features can be used to help distinguish patients with precapillary PH (e.g., PAH) from those with postcapillary PH (e.g., PH-LHD),1,11 distinction of the two conditions can be challenging. For example, mean pulmonary arterial pressure depends on volume load, so it can drop considerably after diuretics.9 Furthermore, accurate PAWP measurement, which can be technically difficult, is very important as there is a cut-off for the identification of pre- and postcapillary PH (≤15 versus >15 mmHg).1
Among patients with severe PH and a clearly dilated right heart, a ventilation/perfusion scan should be performed to rule out chronic thromboembolic PH (CTEPH).10 Such patients often have multiple comorbidities, and approximately 80% have a medical history of pulmonary embolism. Reliable diagnosis of CTEPH is particularly important because there are now various treatment options that can vastly improve pulmonary haemodynamics in these patients.12
A three-step pragmatic approach for the differential diagnosis between Group 1 PH (PAH) and Group 2 PH (mainly HFpEF) has recently been proposed.13 Step 1 is the identification of a specific PH-LHD clinical phenotype: valvular heart disease, LV systolic dysfunction (heart failure with reduced ejection fraction), or LV diastolic dysfunction (HFpEF).13 Of these, PH-HFpEF is the most difficult to distinguish from PAH. Step 2 is to define the pretest probability of PH-LHD.13 Various factors indicate a high versus low probability of PH-LHD, including age >70 versus <60 years, three or more versus no risk factors (obesity, systemic hypertension, dyslipidaemia, impaired glucose tolerance/diabetes), previous versus no previous cardiac intervention, current versus no atrial fibrillation, structural LHD versus no structural LHD, and various parameters measured by electrocardiography, echocardiography, cardiopulmonary exercise testing, or cMRI.13 However, many patients are at intermediate probability, for example, those aged 60–70 years, with 1–2 risk factors, paroxysmal atrial fibrillation, etc.13 Step 3 is haemodynamic assessment of PH-HFpEF.13 If the pretest probability of PH-HFpEF is high, patients should be managed for LHD; if it is intermediate, right heart catheterisation should be considered (for patients with systemic sclerosis, risk factors for CTEPH, or unexplained dyspnoea, but no RV abnormality) or recommended (in case of RV abnormality). If PAWP is >15 mmHg, PH-HFpEF is considered confirmed (intermediate/high probability) or likely (low probability), in which case, LV end-diastolic pressure validation should be considered. If PAWP is 13–15 mmHg, precapillary PH is diagnosed (low probability) or PH-HFpEF cannot be excluded (intermediate/high), in which case provocative testing should be considered.13
The Importance of Imaging in Assessing Right Ventricular Function in Pulmonary Arterial Hypertension
Professor Stephan Rosenkranz
Although PAH is defined by increased pulmonary arterial pressure and vascular resistance, which has to be confirmed by right heart catheterisation, it is the impact of afterload increase on the right heart that is of higher importance.14,15 Initially, structural changes in the heart enable the RV to keep working under strain; however, as PAH progresses, these adaptive changes are insufficient to deliver enough blood to the lungs. Ultimately, this can result in right heart failure, which is the primary cause of death among patients with PAH.14
Because RV structure and function are so important, ESC/European Respiratory Society (ERS) 2015 guidelines recommend regular imaging and haemodynamic assessments.16,17 Along with other factors, these can be used to estimate 1-year mortality risk in PAH (Table 1).16,17 For example, right atrial areas <18 cm2, 18–26 cm2, and >26 cm2 indicate low (<5%), intermediate (5–10%), and high (>10%) 1-year mortality risk, respectively.16,17 In the future, it may be possible to incorporate other imaging parameters into risk prediction models, if data of sufficient quality can be obtained from studies or registries.
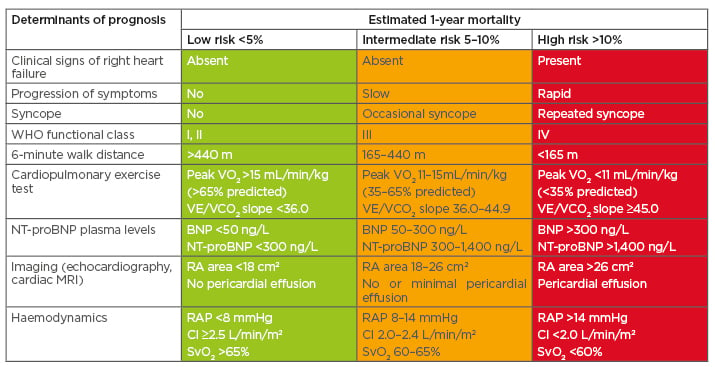
Table 1: Risk assessment in patients with pulmonary arterial hypertension.
BNP: B-type natriuretic peptide; CI: cardiac index; NT-proBNP: N-terminal pro B-type natriuretic peptide; RA: right atrium; RAP: right atrial pressure; SvO2: mixed venous oxygen saturation; VE/VCO2: ventilatory equivalents for carbon dioxide; VO2: oxygen consumption; WHO: World Health Organization.
This material has not been reviewed prior to release; therefore the European Respiratory Society may not be responsible for any errors, omissions or inaccuracies, or for any consequences arising there from, in the content. Reproduced with permission of the © ESC &ERS 2020. European Respiratory Journal Oct 2015, 46 (4) 903-975; DOI: 10.1183/13993003.01032-2015.
Echocardiography is a key imaging tool for diagnosis and follow-up of patients with PH, and can also provide prognostic information.18,19 However, although echocardiography is widely available and can be used to measure a range of RV structural and functional parameters, cMRI is considered the gold standard for a number of important RV parameters.18,20 cMRI can be used to improve risk stratification,21 but is not always available.
Various echocardiographic parameters have prognostic value in PH.18 For example, among 47 patients with PAH, those with tricuspid annular plane systolic excursion ≥1.8 cm had significantly better survival than those with tricuspid annular plane systolic excursion <1.8 cm (approximately 90% versus 45% at 2 years; p=0.009).22 N-terminal pro B-type natriuretic peptide level, a surrogate marker of right heart strain, is also prognostic. For example, in the placebo arm of the Phase III GRIPHON study (n=574), higher levels of N-terminal pro B-type natriuretic peptide (<271 ng/L, 271–1,165 ng/L, and >1,165 ng/L) decreased the chance of remaining free from a morbidity/mortality event (approximately 75%, 50%, and 30%, respectively, at 30 months).23
Various cMRI variables have been correlated with improved survival, including stroke volume index >25 mL/m2, RV end-diastolic volume (RVEDV) index <84 mL/m2, and LV end-diastolic volume index >40 mL/m2 in a study of 64 patients with IPAH.15 In the same study, analysis of the change in various variables after 1-year follow-up showed that mean change in stroke volume index, RVEDV index, and LV end-diastolic volume index predicted mortality.15 Therefore, early diagnosis of PAH and treatment to maintain or improve RV function are important for patient outcome.
In a study of 22 clinically stable patients with IPAH, those with progressive disease had increasing RVEDV and decreasing RV ejection fraction over ≥5-year follow-up, while those with stable disease had little change in RVEDV and increasing RV ejection fraction.6 However, both groups of patients had similar 6-minute walk distance and World Health Organization (WHO) functional class results.6 This indicates that progressive RV dilation precedes clinical worsening.
Overall, improving right heart function in PAH is key to improving patient prognosis,15,24 and these improvements can be measured using noninvasive imaging techniques.
Achieving Best Long-Term Outcomes in Pulmonary Arterial Hypertension: How to Choose the Right Treatments and When to Start Them
Professor Nazzareno Galiè
PAH encompasses a group of rare diseases, with a prevalence of around 50 cases/million.25 Although heterogeneous, there are some common findings, including pulmonary arteriopathy and right heart dilatation.
Various oral therapies are available, which target one of the endothelin pathways (single or dual ERA), the nitric oxide pathway (PDE-5i, soluble guanylate cyclase stimulators), and the prostacyclin pathway (prostacyclin analogues, nonprostanoid prostacyclin receptor agonists).26 The first human epoprostenol (a prostacyclin analogue) study was published in 1984,27 while those for bosentan28 (an ERA) and sildenafil29 (a PDE-5i) were published in 2000. Overall, 41 randomised controlled studies in 9,061 patients with PAH have been carried out: 21 monotherapy versus placebo or versus another monotherapy, 18 monotherapy and/or sequential combination versus placebo, and two initial combination versus monotherapy. The design and endpoints of these studies have evolved over time, from small, short studies of exercise capacity30-39 to larger, longer outcome trials.40-43 Between 1998 and 2015, the number of ESC/ERS 2015 guideline-approved PAH drugs steadily increased, from one to 11, but the number of pathway classes has remained constant at three since 2003.16 Drugs that have been approved according to the ESC/ERS 2015 PH guidelines16,17 are ERA (ambrisentan, bosentan, macitentan), PDE-5i (sildenafil, tadalafil), a soluble guanylate cyclase stimulator (riociguat), and prostacyclin analogues (iloprost, treprostinil, epoprostenol); and more recently, a prostacyclin receptor, also termed IP, agonist (selexipag).44
Early trials tested monotherapies, but then studies moved on to test sequential combinations, and then initial combinations. In the SERAPHIN study, 742 patients with PAH were randomised to macitentan 3 mg, 10 mg, or placebo.41 Among 308 patients who were already receiving background PAH therapy (mainly [97.4%] a PDE-5i) at baseline and were randomised to placebo or macitentan 10 mg (i.e., sequential combination), the addition of macitentan reduced the risk of a morbidity/mortality event by 38% (hazard ratio [HR]: 0.62; 95% confidence interval [CI]: 0.43–0.89; p=0.009).45
In the GRIPHON trial, 1,156 patients with PAH were randomised to selexipag (the only oral treatment that targets the prostacyclin pathway that is approved in Europe) or placebo.42 Among 376 patients who were already receiving a PDE-5i and an ERA, the addition of selexipag (i.e., third drug in sequential combination) was associated with a 37% reduction in morbidity/mortality when compared with double combination therapy (HR: 0.63; 95% CI: 0.44–0.90).46 This beneficial effect was more pronounced among patients with mild symptoms (WHO functional Class II; n=115) (64% risk reduction; HR: 0.36; 95% CI: 0.14–0.91) than among those with more advanced symptoms (WHO functional Class III; n=255) (26% risk reduction; HR: 0.74; 95% CI: 0.50–1.10),46 highlighting the importance of early intervention to delay disease progression.
In the AMBITION study, 500 treatment-naïve patients with PAH were randomised 1:1:2 to ambrisentan, tadalafil, or both (i.e., initial combination).43 Those who started on combination therapy had a 50% lower risk of a morbidity/mortality endpoint than the pooled monotherapy population (HR: 0.50; 95% CI: 0.35–0.72; p<0.001).
Macitentan plus tadalafil has been tested as an initial combination therapy in 46 treatment-naïve patients with PAH in the single-arm OPTIMA study.47 PVR (primary endpoint) was reduced from 11.7±4.7 WU at baseline to 6.5±3.6 at 16 weeks (47% reduction; geometric mean ratio: 0.53; 95% CI: 0.47–0.59; p<0.0001). Six-minute walk distance (a secondary endpoint) also improved, from 352±135 m to 388±142 m (+36 m; 95% CI: 16–56; p=0.0008).47
In the most recent ESC/ERS guidelines,16 patients with PAH can be stratified into estimated 1-year mortality risk categories (low <5%, intermediate 5–10%, high >10%) based on various factors. These risk levels can then be used to assign patients to an appropriate treatment option,48 as discussed in more detail below, which is key to achieving the best long-term outcomes.
The ESC/ERS risk stratification has been validated in two retrospective registries.49,50 The Swedish PAH registry (n=530) reported 5-year survival rates of approximately 84%, 52%, and 35% for low-, intermediate-, and high-risk patients, respectively,49 which was similar to the COMPERA Registry (n=1,588) (approximately 77%, 53%, and 33%, respectively).50 The French PAH registry reported decreasing transplant-free survival with decreasing numbers of low-risk criteria.51
The ESC/ERS guidelines recommend that PAH severity is assessed at diagnosis based on clinical assessment, exercise tests, biochemical markers, and echocardiographic and haemodynamic evaluations; with regular follow-up assessments every 3–6 months in stable patients (both Class I, level C).16,17 In terms of treatment, they recommend the achievement/maintenance of a low-risk profile (Class I, level C). All these recommendations will be elevated to level B in the new guidelines, which are planned for 2022.
According to the 6th World Symposium on Pulmonary Hypertension (WSPH) 2018 treatment algorithm48 and the 2015 ESC/ERS guidelines,16,17 vasoreactive patients should be treated with calcium channel blockers. Nonvasoreactive patients should be treated according to risk: high-risk patients should receive an initial combination therapy including intravenous prostacyclin analogue, while intermediate/low-risk patients should be given an initial oral combination, although initial monotherapy currently has a residual role (e.g., PAH and age >75 years; suspicion/high probability of pulmonary veno-occlusive disease; HIV; portal hypertension; very mild disease; or haemodynamic responders).48 After 3–6 months of treatment, risk should be reassessed. If patients are low risk, their treatment should be continued; if they are intermediate/high risk, a triple sequential combination is recommended. If patients remain at intermediate/high risk after a further 3–6 months, maximal medical therapy and listing for lung transplantation are recommended.48
Closing Remarks
Doctor Irene Lang
Firstly, it is essential to differentiate pre- from postcapillary PH because vasodilators work best in precapillary disease. Secondly, although PAH is a pulmonary vascular disorder, it is RV function that determines survival. Thirdly, vasodilator treatments have improved 3-year survival rates from 30–40% to >85%.52 Fourthly, treatment regimens are based on regular risk assessments (every 3–6 months). Lastly, the use of upfront combination medications is beneficial.
Date of preparation: November 2020
EM-41594