Abstract
A biomarker is any measurement taken that aims to improve a diagnosis, or predict the response, to treatment of disease. Although not limited to laboratory molecular markers, this variety have attracted the most interest and seen the greatest development in recent years. The field of cardiology was an early adopter of biomarkers, with transaminases having been used for the diagnosis of acute myocardial infarction since the 1970s. The use of biomarkers has become increasingly prevalent since then and provided ever more sensitive means to diagnose myocardial cell injury or heart failure. However, diagnosis of disease at an increasingly earlier stage leads to blurring of the line between health and disease and we may be reaching the limits of early detection. Biomarkers may evolve to provide a greater understanding of the pathogenesis of cardiac disease, and by extension, the differentiation of disease subtypes. This article will review the evolution of cardiovascular biomarkers, the advantages and pitfalls associated with their use, as well as the future direction of cardiac biomarker research.
INTRODUCTION
The use of biomarkers has become routine in many areas of medicine. Knowing how and why these became incorporated into medical practice allows us to use them to their maximum potential and helps guide future developments. Biomarkers are especially widespread in the field of cardiology where disease may manifest with subtle or no clinical signs. This narrative review covers the evolution and current application of biomarkers for acute coronary syndromes (ACS) and heart failure (HF). Pitfalls in their use and areas of cardiology poorly served by existing biomarkers will be discussed. This review will not cover biomarkers used for cardiovascular risk prediction, including lipoproteins or homocysteine, nor more novel work regarding single nucleotide polymorphisms that may confer risk of coronary artery disease, which are covered elsewhere.1-3
SEARCH METHODOLOGY
The authors conducted a comprehensive search in the English-language literature to identify relevant studies, regardless of publication status or year of publication. PubMed and Google scholar databases were searched combining the terms ‘cardiovascular’ OR ‘cardiac’ OR ‘myocardial’ OR ‘heart’ OR ‘heart failure’ OR ‘myocardial infarction’ OR ‘AMI’ AND ‘biomarker’ OR ‘circulating marker’ OR ‘cardiac protein’. Studies could include early or late phase human trials. Backward (using reference list of paper) and forward (using studies citing a paper) snowballing were applied to identify further studies. An extensive list was developed, and a shortlist was created based on the limitations of the length of the narrative review and importance of the marker. A separate search was also done for papers relating to the individual markers in PubMed. The last search was performed in January 2020.
WHAT IS A BIOMARKER?
Definition of a Biomarker
A biomarker may be defined as ‘a characteristic that is objectively measured and evaluated to aid in understanding one or more of: the prediction of disease, its cause, the diagnosis, and the response to intervention.’4 Under this definition, even commonplace parameters such as blood pressure and heart rate are biomarkers. However, the modern conception relates to clinical biochemistry.
History of Cardiac Biomarkers
The first recorded example of a cardiac biomarker came from an observation in 1954 detailing a rise in blood aspartate aminotransferase concentration 3–4 hours after an acute myocardial infarction (AMI).5 The following year, a similar increase in serum lactate dehydrogenase activity was also seen post-AMI.5 Thereafter, creatine kinase (CK) was found to be far more sensitive than aspartate aminotransferase or lactate dehydrogenase, with ≤98% sensitivity within 72 hours.6 However, these enzymes exist in skeletal as well as cardiac muscle, leading to many false positives. This problem was partly overcome by use of an isoenzyme of CK called CK-myocardial band (MB), constituting a far greater proportion in the myocardium despite also being present in skeletal muscle. The separation of serum isoforms of CK isoenzymes using polyacrylamide gel electrophoresis, thereby identifying the MB of CK, was first described in 19727 and heralded the ubiquity of use through the 1980s.
Measurement of peptides and proteins in biological fluid was revolutionised through the development of the radio-immunoassay by Berson and Yallow in late 1960s. Radio-immunoassays allowed for the precise and accurate measurement of tiny quantities of proteins, evident from the successful measurement of myoglobin in 1978.8 However, the concerns of experimental complexity and radiation use led to the replacement of the radiolabel with enzyme labels in the immunoassay: the ELISA was born.9 CK-MB mass was first measured by ELISA in 1985, replacing the more laborious electrophoretic method.10
In 1985, the Thrombolysis in Myocardial Infarction (TIMI) study group reported that patients with AMI received maximum benefits when receiving thrombolysis within 4–6 hours of chest pain.11 Therefore, an early biochemical marker of AMI became crucial for the diagnosis of myocardial injury. This helped to promote the development of the troponin (Tn) assay.
TROPONIN
Tn are part of the thin filament in the sarcomere and are involved in the calcium-dependent interaction of actin and myosin. Tn are found in striated muscle; however, subunits T and I are cardio-specific (cTn). Tn release from the myocardium may occur as a result of normal cellular turnover (Figure 1):12 apoptosis; cellular release of cTn degradation products; increased (reversible) cellular wall permeability; swelling of cardiomyocytes, leading to formation and release of membranous ‘blebs’; and myocyte necrosis.13
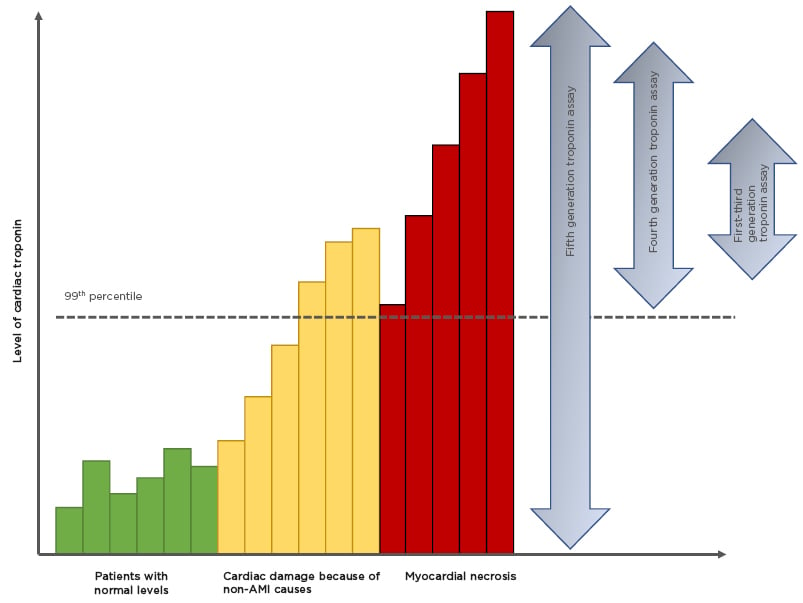
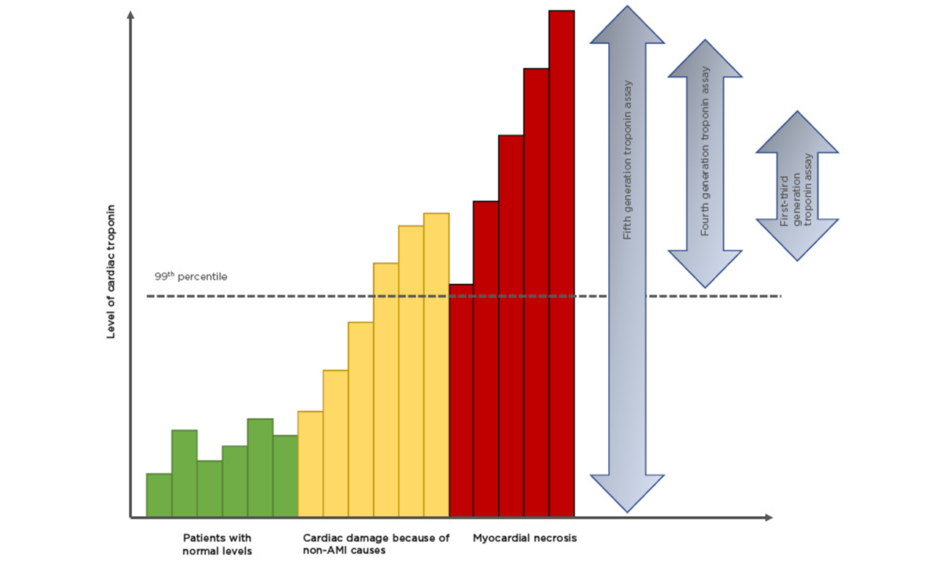
Figure 1: Improving sensitivities of troponin assays.
There is a background, normal, turnover of cardiac troponin (green bars). With the onset of AMI there is a rise in cardiac troponin that represents either ischemia-induced release of cytosolic troponin or micro-necrosis (orange bars). Between 2 and 6 hours there is a steep increase in cardiac troponin representing myocardial necrosis (red bars).
Modified from Garg et al.12
AMI: acute myocardial infarction.
In the late 1990s it was shown in patients who were Tn-positive (but CK-MB-negative) that early medical intervention significantly improved outcomes. This led to the redefinition of AMI in the year 2000 to use cTn instead of cardiac enzymes or CK-MB mass.14
High-Sensitivity Cardiac-Specific Troponin
High-sensitivity (hs)cTn assays are designated by their ability to detect cTn even in healthy individuals.15 By definition these assays must be able to measure cTn concentrations in >50% of a healthy reference population (hence 50% of the population are above the lower limit of detection).13 Therefore, ‘high-sensitivity’ refers to analytical and not clinical sensitivity. The latest generation of hscTn assays can detect troponin in >95% of a reference population.16 Such high sensitivity is at the cost of low specificity, i.e., the ability to detect individuals without disease. The upper limit of the reference interval is considered to be the 99th percentile of the derivation population and assays must have a coefficient of variation (CV) <10% at that point.17 Until recently this had been unachievable, but with advances in the immunoassay techniques the low CV has been met. The fifth generation hscTn assay is capable of measuring levels as low as 5 ng/L,18 blurring the line between health and disease (Figure 1).12 Almost 1 in 100 of the host population would have a raised hscTn: therefore how do we differentiate ‘normal’ from ‘diseased’?19
Perhaps surprisingly it is acceptable to calculate the 99th percentile (per sex) using as few as 300 males and 300 females.20 Several factors are known to positively skew a troponin result, including age, male sex, low glomerular filtration rate (GFR), reduced left ventricular function, and systemic inflammation.21
The cut-off value by the 99th percentile rule will only be clinically useful when applied to patients with a high pre-test probability of ACS. This introduces the concept of Bayesian reasoning,22 i.e., the diagnosis of AMI should always be made only after consideration of the patient history and 12-lead echocardiography, with hscTn concentrations interpreted within a well-defined and validated algorithm or pathway.23,24
The origin of the Tn assay was to determine whether a patient was having a Type 1 myocardial infarction, caused by coronary plaque rupture or erosion (Table 1). As assay sensitivity has improved, elevations caused by other pathologies are observed (such as Type 2 myocardial infarction; Figure 1).12 This decreases the positive predictive value of a solitary Tn elevation for acute atherothrombotic coronary artery disease.25 Changes in cTn values can be used to distinguish acute from chronic disease. Absolute changes are assay-dependent but appear superior to relative (percentage) changes using hscTn assays. High levels are associated with detrimental outcomes. The precision and reproducibility of hscTn assays allows for accurate, serial measurements in a short time. The value of the hscTn assay is not to identify more AMI, but to more quickly identify patients without disease.26 Because of the low CV of the latest hscTn, the wait for a repeat test has fallen from 6–12 hours to 1 hour after symptom onset.27 The European Society of Cardiology (ESC) recommends the ‘0/1h-approach’ only if using hscTn with a validated algorithm in patients presenting >3 hours after chest pain onset; otherwise, the 0/3hr testing algorithm is recommended.24 New point-of-care hscTn tests are becoming available, with high diagnostic accuracy.16 These will facilitate the implementation of algorithms demanding fast decision making.
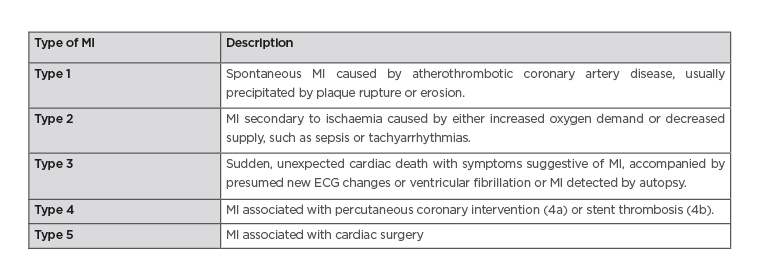
Table 1: The fourth universal definition of myocardial infarction.
ECG: echocardiogram; MI: myocardial infarction.
Adapted from Thygesen et al.13
The high sensitivity of hscTn means that the positive predictive value for AMI ranges from as low as 15% to up to 75%.28 Many individuals will undergo further assessment, potentially delaying treatment of noncardiac causes of an elevated hscTn.29 Is the increase in downstream testing worth it? Evidence suggests that hscTn improves the early diagnosis of AMI and risk stratification, and outcomes have improved following hospitalisation for AMI.30 However, high sensitivity also leads to the epidemiological paradox called the ‘Will Rogers phenomenon’, in which individuals formerly considered ‘healthy’ are now considered ‘ill’. Regardless, these individuals are not as ill as patients diagnosed using older assays. Reclassification may therefore result in AMI having a ‘better’ outcome.
Future Tn research may be directed at development and validation of algorithms for estimation of the pre-test probability of ACS, as well as determination of optimal post-test probability thresholds for initiating therapy.22
NOVEL BIOMARKERS UNDER EVALUATION FOR ACUTE CORONARY SYNDROME
The rationales for newer biomarkers in ACS are to improve the prognostic performance of Tn; reveal the pathophysiology of myocardial ischaemia (improved specificity); or to instantly rule out AMI, without the need for additional blood draw (improved sensitivity). Many candidates have been proposed, but for brevity the most salient are discussed. These include early biomarkers of cardiomyocyte injury such as H-FABP and cMyBP-C; markers of neurohormonal activation, including copeptin and MR-proADM; and markers of vascular inflammation such as C-reactive protein (CRP) and MPO.
Copeptin is the C-terminal end of the vasopressin prohormone, co-released with arginine vasopressin within 0–4 hours of AMI symptom onset.31 Arginine vasopressin is a stress hormone and has a short half-life, meaning copeptin acts as a surrogate marker. Because endogenous stress is invariably present at early presentation of AMI, copeptin may help to improve the sensitivity of cTn.23 A similar conception underlies the use of MR-proADM. This is a more stable form of the adrenal stress hormone adrenomedullin. Elevated levels have been shown in AMI but has yet to clearly add prognostic value above established methods.32
The sarcomeric protein cMyBP-C is twice as abundant in the heart as cTn or cardiac-specific troponin 1 (cTnI) and is a specific marker of myocyte injury.28 It is released more rapidly than cTn and may be superior for those experiencing chest pain for <3 hours.29,33 Similarly, H-FABP is another intra-cardiomyocyte protein also proposed as a sensitive, early marker of myocellular injury; however, its incremental value to cTn has not been established.34,35
Acute cardiac injury induces myocardial infiltration of leukocytes and proliferation of fibroblasts.2 Pro-inflammatory cytokines like IL-6 and acute-phase proteins such as CRP are upregulated in patients with AMI and the degree of inflammatory response has been linked to risk stratification.2 MPO is an enzyme released by neutrophil degranulation and contributes to inflammation, thereby worsening cardiac remodelling and having long-term adverse cardiac sequelae.2,36
A multi-marker strategy combining biomarkers representing the components outlined above (myocardial stress, myocyte necrosis, and inflammation) may provide additive prognostic information.36
NATRIURETIC PEPTIDES
The clinical diagnosis of HF can be challenging.37 Several clinical criteria exist (Framingham, National Health and Nutrition Examination Survey [NHANES], modified Boston, Gothenburg, and International Classification of Disease 9th Revision). The Framingham clinical criteria are the most sensitive (90–92%), but with 40–79% specificity.38,39 A biomarker to aid in the diagnosis of HF was therefore welcomed following the discovery of an ‘atrial natriuretic factor’ in the early 1980s.40
Natriuretic peptides (NP) comprise atrial natriuretic peptide (ANP; formerly known as atrial natriuretic factor), brain natriuretic peptide (BNP), C-type natriuretic peptide, and urodilatin. The first discovery of this family of peptides was made by de Bold in 1981,38 who showed that extracts of rat atrium contained a substance that increased salt and urine output in the kidney. BNP is a 32-amino acid peptide, structurally similar to ANP, with a common 17-amino acid sequence. It was isolated in 1988 from porcine brain extracts by Sudoh et al.41 BNP is a misnomer as it is synthesised and released primarily from ventricular myocardium.42 Cleavage of the prohormone proBNP produces the biologically active 32-amino acid BNP, as well as biologically inactive N-terminal proBNP (NT-proBNP; 76 amino acids). Both are released from ventricular cardiomyocytes in response to mechanical stretch42 and proportionate to the severity of HF.42-44 In the ‘Breathing Not Properly‘ study a threshold BNP of 100 pg/mL was more accurate for HF (83%) than either the NHANES criteria (67%) or the Framingham criteria (73%).45 The optimal diagnosis of HF was made when BNP was used in conjunction with other clinical information.
Ventricular stretch may occur as a result of renal failure, just as much as HF. In pre-dialysis patients, the estimated GFR and left ventricular mass index correlated independently to plasma BNP and NT-proBNP concentrations.46 Unlike NT-proBNP, which relies solely on renal clearance, plasma BNP is also cleared by endopeptidases and receptor capture, and so values are less dependent on GFR. BNP may therefore be more appropriate for use in chronic kidney disease.46
The newly licenced Sacubitril/Valsartan is the first agent in a new class of angiotensin receptor neprilysin inhibitors. Inhibition of neprilysin prolongs the half-life of ANP and BNP, allowing for greater vasodilatory and natriuresis effects.47 Use of this medication will mean persistence of elevated BNP, whereas NT-proBNP level will continue to be reflective of volume status. Related to this is the question of whether HF medications may be titrated against NP; this was addressed by the GUIDE-IT trial, which suggested titration against NP was no more effective than a usual clinical strategy.48 Conversely, a more recent meta-analysis has suggested a benefit.49 The dissimilarity may reflect the approach to management in the control groups, more than the intervention.
The short half-life of ANP (2–5 minutes) has made it unsuitable as a biomarker. However, newer assays can measure mid-regional proANP (MR-proANP) which is the precursor hormone of ANP and more stable. MR-proANP may improve diagnostic certainty in acute HF when there is associated obesity or chronic kidney disease, and also more accurately predicts 90-day mortality than NT-proBNP.50 There is insufficient evidence for MR-proANP to replace the clinical utility or BNP, although it is now included in the ESC guidelines.51 These guidelines also recommend using cut-off values for diagnosing HF (BNP >35 pg/mL; NTpro-BNP >125 ng/mL) in stable patients. However, higher value BNP >100 pg/mL, or NT-proBNP >300 pg/mL, should be used in the acute setting.51
PROBLEMS WITH EXISTING BIOMARKERS IN HEART FAILURE
Stimuli for natriuretic release include myocardial ischaemia (even in the absence of necrosis),52 and hypoxia via HIF.53 The physiological considerations discussed above explain why factors including blood pressure,43 kidney function,54 anaemia,55 and ageing56 will affect circulating BNP directly. Extra-cardiac factors, e.g., sex or obesity, are also associated with elevated BNP.56,57 Conversely, almost a quarter of patients with chronic symptomatic HF may have NP levels in the lower ranges.58 HF is a complex clinical syndrome caused by progressive structural and mechanical dysfunction, which gives rise to a multifactorial, interlinked pathophysiology (Figure 2). This makes aetiological certainty difficult and currently we do not have biomarkers that allow for an individualised diagnosis or response to therapy. This may be particularly problematic in those with heart failure with preserved ejection fraction, who are already disadvantaged by a lack of treatment modalities with proven mortality benefit.
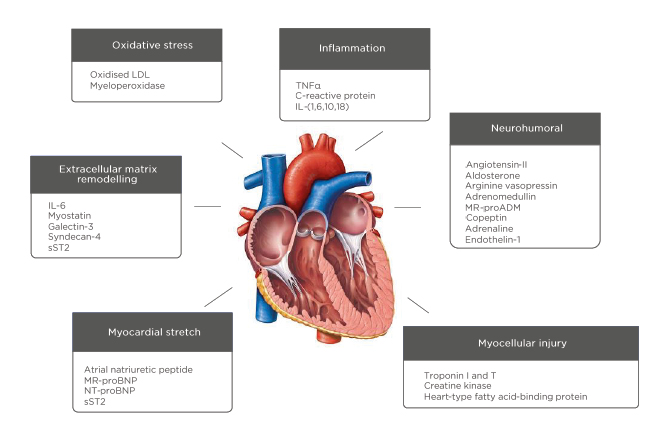
Figure 2: The pathophysiology of heart failure and potential biomarkers.
LDL: low density lipoprotein; MR-proADM: mid-regional pro-adrenomedullin; MR-proBNP: mid-regional-pro B-type natriuretic peptide; NT-proBNP: N-terminal pro B-type natriuretic peptide; sST2: soluble suppression of tumorigenesis-2.
FUTURE CARDIAC BIOMARKERS IN HEART FAILURE
As illustrated (Figure 2), there is a multitude of new biomarkers under evaluation representing pathophysiological pathways including inflammation, oxidative stress, and fibrosis. Candidate markers include cytokines, peptides, proteins, metabolites, and circulating nucleic acids. The success or failure of these markers for case selection will be dictated not just by their accuracy and reliability, but by whether specific anti-inflammatory, anti-oxidant, and anti-fibrotic therapies become established.
Cardiomyocyte Remodelling
Fibrous tissue deposition in the cardiac interstitium can result from many types of cardiac injury and is associated with increased disease severity and adverse outcomes.59,60
ST2, also known as IL1-RL1, is a member of the Toll-like/IL-1 receptor superfamily. It is upregulated by mechanical stress of cardiomyocytes and cardiac fibroblasts.
The ligand for ST2 is IL-33, and binding leads to a cardioprotective effect by inhibition of the Th2-dependent inflammatory response. However, soluble ST2 (sST2) circulating in plasma sequesters IL-33, meaning that the heart is more exposed to fibrotic change. sST2 levels are an independent predictor of mortality and HF hospitalisation in patients with acute or chronic HF, with an additive prognostic value to NP.61 Better control of HF lowers sST2. Importantly, sST2 is relatively unaffected by age, sex, obesity, aetiology of HF, atrial fibrillation, and anaemia.62
Galectin-3 is a member of the lectin family secreted by activated macrophages. It may complement other biomarkers by providing an upstream signal in the fibrotic process. Given that cardiac fibrosis is irreversible, this provides a timely measure of risk to the myocardium. Galectin-3 is not acutely changed by cardiac decompensation, nor is it acutely responsive to standard therapies for HF. It is therefore less effective than NT-proBNP for diagnosing acute HF,63 but nonetheless aids prognostication; subsequent to the DEAL-HF study, it was found that participants with elevated galectin-3 and NT-proBNP had worse survival than those with either marker elevated alone. However, the value of Galectin-3 for HF prognosis may diminish after adjusting for renal function.64
Inflammation
The association of high-sensitivity CRP with cardiovascular risk has been known for over two decades.65 However, recent work suggests it may directly influence the inflammatory processes contributing to myocardial damage. In animal models, CRP removal after ST segment-elevated myocardial infarction reduced infarct size.66 CAM1 is an ongoing trial looking at the benefit of eliminating CRP from serum in ST segment-elevated myocardial infarction. Preliminary results show a reduction of infarct size and improved ventricular wall motion.67
Multi-Marker Panels
Drawing biomarkers together in multi-marker prediction models has the potential to improve risk stratification. For example, the plaque disruption index consists of MPO, hsIL-6, MRP8/14, and PAPP-A, and is being tested to identify Type 1 AMI caused by plaque rupture (Table 1). In a pilot study, the plaque disruption index had better diagnostic accuracy than coronary angiography.68 For HF, a panel including MR-proADM, hscTn, cFLC, hsCRP, and ST2 added prognostic value to standard measures of HF, whereas, individually, each added little.69
Multi-Omics
Genetic information is transcribed from DNA into RNA and translated into proteins, which can then produce metabolites. These components may be evaluated by genomics, transcriptomics, proteomics, and metabolomics, respectively. Measurement was once a laborious process but may now be done at scale and at speed. This leads to a vast amount of personalised data that needs to be integrated to facilitate diagnosis and provide a unique risk assessment and personalised treatment. In an early example of such an approach using mass spectrometry profiling, Cheng et al.70 demonstrated that a metabolomic panel of histidine, phenylalanine, spermidine, arginine, and phosphatidylcholine C34:4 had higher prognostic value for the combined endpoints of death and HF-related hospitalisation than BNP.70
Micro RNA (miRNA) are small non-coding RNA molecules that act as post-transcriptional regulators of gene expression.2 They can be released into the circulation, where they may be found attached to proteins or in extracellular vesicles. Increases or decreases of >30 miRNA may orchestrate changes to the transcriptome, and ultimately the proteome, during HF. This may, for example, lead to differentially expressed proteins involved in glycolysis, β-oxidation, and ketone metabolism in the failing heart,72 as well as promote the development of cardiac fibrosis, e.g., as caused by miRNA‑21.
Hower, the integrative analysis of ‘omics’ data are not straightforward and represents many logistical and computational challenges. Machine learning may be required to create clinically useful diagnostic panels.72
CONCLUSIONS
The last 70 years have witnessed huge progression in the development and sophistication of laboratory biomarkers: from laboriously measured single protein analyses, to multi-array platforms and miRNA. The future generation of biomarkers promise greater diagnostic and prognostic accuracy, as well as allowing for targeted therapy and measures of treatment response. However, the central tenet is likely to remain: that biomarkers are most useful when applied to a specific clinical question, and not before.