Abstract
Acute coronary syndrome (ACS) encompasses a spectrum of clinical disorders of myocardial ischaemia or infarction, with atherosclerosis leading to coronary plaque formation the predominant disease process. Alterations of endothelial cell integrity involving atherosclerotic plaque surfaces, such as plaque rupture or erosion, can lead to atherothrombosis with subsequent interruption to myocardial blood supply. Over the past two decades, it has become increasingly apparent that inflammation plays a pivotal role in the initiation and progression of atherosclerosis. Inflammatory cytokines have been shown to correlate with the risk and burden of coronary artery disease and there is a growing body of evidence demonstrating the presence of various immune cells in atherosclerotic plaques and coronary thrombus specimens. Due to improved cellular detection methods compared to earlier studies, neutrophils are being increasingly recognised as a key player in the process of athero-inflammation. The aim of this review is to: i) outline the role of neutrophils in ACS and atherothrombosis, ii) describe the process of inflammasome-mediated release of inflammatory cytokines from neutrophils, and iii) discuss multiple parameters of neutrophil activity in ACS, including peripheral neutrophil/lymphocyte ratio; neutrophil microparticle release; expression of neutrophilic granular proteins, including myeloperoxidase, neutrophil elastase, and metalloproteinases; neutrophil extracellular traps release; tissue factor; and neutrophil-macrophage interactions.
INTRODUCTION
Acute coronary syndrome (ACS) encompasses a spectrum of clinical disorders of myocardial ischaemia or infarction, presenting as unstable angina, non-ST segment elevation myocardial infarction (NSTEMI) or ST-segment elevation myocardial infarction (STEMI). There are various aetiologies underpinning ACS, but the predominant disease mechanism is atherosclerosis, with the potential for subsequent plaque disruption leading to thrombosis and cessation of myocardial blood flow.
Atherosclerosis is a progressive disease of plaque deposition, which consists of lipids, smooth muscle cells, and calcium within the layers of the blood vessel. It is thought to be triggered by the deposition of cholesterol-containing plasma lipoproteins in the subendothelium at points of altered flow haemodynamics, leading to inflammatory changes in endothelial cells.1 Over time, there is gradual narrowing of the lumen of the coronary artery, leading to myocardial ischaemia as well as disruption of laminar blood flow, further disrupting the integrity of endothelial cells. Disruption of coronary plaque and resultant exposure of its contents (lipids, necrotic core containing inflammatory and apoptotic cells) causes platelet aggregation and thrombosis. If complete occlusion of the coronary blood supply occurs, this results in a STEMI, whereas a NSTEMI results from a transient or incomplete obstruction of flow.2 These mechanisms have been extensively researched in clinicopathological studies and a spectrum of plaque surface alterations have been identified, including fissuring, erosion, ulceration, and rupture.3 The mechanism of disruption of atherosclerotic plaques with the highest risk of atherothrombosis have broadly been classified into plaque rupture and plaque erosion. Plaque rupture is defined as a structural defect in the fibrous cap that separates the lipid-rich necrotic core of a plaque from the lumen of the artery with destruction of the media layer, whereas plaque erosion is when the endothelium is missing but the media layer remains intact. These two processes of plaque remodelling differ phenotypically in terms of cellular content, inflammatory activity, and extracellular matrix composition, as evidenced by autopsy findings of patients with ACS.4 Ruptured plaques are associated with larger necrotic cores, higher macrophage content, but decreased smooth muscle cells and extracellular matrix (hyaluronic acid, proteoglycans) compared to eroded plaques. Intravascular imaging techniques, such as optical coherence tomography, are now able to identify thin cap fibroatheromas and assess necrotic core thickness, both of which are associated with plaque rupture.5
There is convincing evidence from animal studies, that in addition to plaque characteristics, the immune system and inflammation have a significant role in atherosclerosis and the pathogenesis of ACS. Modified lipoproteins trapped within the subendothelium attract circulating inflammatory cells, which then infiltrate into atherosclerotic plaque. Various immune cells, including lymphocyte subsets, dendritic cells, platelets, and monocytes/macrophages, have been implicated in atherosclerosis-associated inflammation. However, there is now growing evidence that neutrophils play a key role in linking the immune system and atherosclerosis. Neutrophils are the most abundant white blood cell in humans and are an important component of the innate immune system, which is the non-specific first line of defence against pathogens. Neutrophils contain a variety of granular proteins, including antimicrobial peptides, proteases, and reactive oxygen species (ROS), which are released to destroy offending pathogens.6 Neutrophil production is primarily regulated by granulocyte colony stimulating factor (G-CSF) and interleukin (IL)-23 whilst retention of neutrophils in the bone marrow is regulated by chemokine receptors CXCR2 and CXCR4. Hyperlipidaemia can lead to dysregulation of neutrophil homeostasis and promotes mobilisation of neutrophils from the bone marrow as well as stimulation of granulopoiesis in the bone marrow and spleen.7 Neutrophils are primed by either external stimuli (foreign pathogens) or internal stimuli (cytokines and chemokines), which guide the primed neutrophils to the site of infection or inflammation. This then leads to the mobilisation of intracellular granules that contain preformed receptors to the plasma membranes and results in the expression of receptors (intra and extracellular) and cytokines, which further enhance neutrophil function. Neutrophil granules, which contain lysosomal proteins such as myeloperoxidase (MPO), neutrophil elastase, cathepsin G, protease-3, and matrix metalloproteinases (MMP) are mobilised upon priming of the cell.
In atherosclerosis, failed attempts to clear lipoproteins from the subendothelium of blood vessels and the death of immune cells (mainly macrophages) and smooth muscle cells lead to the formation of a proinflammatory necrotic core. In response to this, local inflammatory cells within the coronary vasculature, such as endothelial cells, leukocytes, and platelets, generate and release cytokines, small proteins involved in cell signalling. These cytokines have the potential to activate humoral and cellular factors causing disruption of the balance of anticoagulants and procoagulants, leading to downstream activation of the complement and coagulation pathways, which leads to inflammation and thrombotic processes. This is reflected in multiple clinical studies demonstrating a strong association between multiple inflammatory biomarkers (hsCRP, IL-6) and the risk of incident coronary artery disease.8,9 The inflammasome is a multiprotein immunomodulatory complex and consists of many different subtypes. Of these, the NLRP3 inflammasome has been extensively studied and plays a significant role in athero-inflammation.10 The activation of the NLRP3 inflammasome in neutrophils activates caspase-1, which is responsible for the generation of inflammatory cytokines, including IL-1β and IL-18, both of which have been implicated in coronary artery disease and atherothrombosis.11,12 Preliminary data from the CANTOS trial, which investigated the efficacy of canakinumab, an IL-1β antagonist, in patients with a history of myocardial infarction and a persistently elevated CRP level despite contemporary secondary prevention strategies, has shown a decrease in the recurrence of major adverse cardiovascular events.13 Trans-coronary (difference between systemic and coronary sinus) IL-1β and IL-18 levels have also been shown to strongly correlate with disease activity in patients with ACS.14 Moreover, a reduction in inflammatory cytokines with statin treatment has been shown to reduce atherosclerosis progression on intracoronary imaging, as well as cardiovascular events, including death and myocardial infarction in patients with ACS, independent of atherogenic lipid levels.15
The role of neutrophils in atherosclerosis was largely unappreciated in initial studies due to their limited life span, high turnover with rapid clearance by macrophages, marked phenotypic variation of neutrophil surface markers, and a lack of adequately sensitive and specific detection methods.7,16 More recently however, their identification in atherosclerotic plaques has increased due to better cellular detection methods. Neutrophils are now able to be identified in tissue specimens thanks to improved live imaging fluorescent intravital microscopy17 techniques, such as multi-photon excitation imaging.18 This is used in conjunction with immunohistochemistry, enzyme-linked immunosorbent assay (ELISA), and flow cytometry methods using antibodies against lymphocyte antigen 6 complex locus G6D, Ly6G (expressed on mouse neutrophils), and human neutrophil cell surface markers CD-66b, CD-177, and CD-11b, as well as neutrophilic proteins such as myeloperoxidase (MPO) and elastase. Neutrophils, identified on intravital and confocal microscopy using flow cytometry with staining against Ly6G and leukocyte common antigen CD45, along with enhanced green fluorescence protein detection, have been demonstrated in the shoulder regions of atherosclerotic plaques from enzymatically digested mouse aortas.19 Another mouse model showed that specific neutrophil depletion led to a reduction in atherosclerotic plaque sizes in mice who were given a high fat diet.20 In addition to animal studies, CD66b-positive neutrophils have also been identified in human autopsy atherectomy specimens, coronary thrombi, and peripheral blood samples from patients with ACS.21-23
NEUTROPHIL EFFECTS ON MONOCYTE/MACROPHAGE FUNCTION
Neutrophils are important facilitators of monocyte/macrophage function. Monocytes migrate into the subendothelial space, ingest unchanged and oxidised lipoproteins, and then transform into cholesterol-laden foam cells. These foam cells contribute to a proinflammatory state within advanced atherosclerotic plaques and also form necrotic cores that can cause plaque destabilisation and rupture.24 Neutrophils within atherosclerotic plaques play a role in monocyte recruitment and adhesion in early atherosclerosis via monocyte chemoattractant protein-1 (MCP-1), azurocidin, as well as granule proteins cathepsin G and LL-37/ cathelicidin.25 In addition to recruitment, circulating neutrophils also play a role in the activation of monocytes via toll-like receptor (TLR)-4 and the generation of soluble IL-6 receptors.26 Neutrophils have been shown to effect efferocytosis, the process of removing dying cells and components of the necrotic core within plaques, by releasing high mobility group box 1, which binds to integrins on macrophages and inhibits MFG-E8, a bridging molecule between apoptotic cells and macrophages during efferocytosis.7 Defective efferocytosis leads to an accumulation of necrotic cells, resulting in persistent plaque inflammation as well as increasing the size of the necrotic core, both of which are associated with plaque rupture.27
NEUTROPHILS IN PLAQUE REMODELLING
As mentioned earlier, alterations and remodelling of atherosclerotic plaque surfaces can lead to superimposed thrombosis and clinical events. Neutrophils play a significant role in the process of atherosclerotic plaque erosion, which has been estimated to underlie ~40% of coronary atherothrombotic events.28 TLR are pattern recognition proteins that are key components of the innate immune system. TLR are found on many cells, including coronary endothelial cells, and their agonism leads to multiple important cellular mechanisms, including the generation of endoplasmic reticulum stress-ROS, endothelial cell apoptosis, and impairment of endothelial cell repair. This causes detachment of plaque endothelial cells, leading to superficial erosion and thrombotic complications. Circulating and intra-plaque neutrophils potentiate and amplify effects of TLR2 agonism, causing the propagation of superficial plaque erosion.29 A murine model of plaque erosion demonstrated substantial neutrophil accumulation at sites of early erosion as a result of endogenous TLR2 activation of endothelial cells.30 This is clinically relevant, with autopsy studies showing an increased proportion of ACS cases to be caused by plaque erosion compared to overt plaque rupture.4
PARAMETERS OF NEUTROPHIL ACTIVATION AND FUNCTION IN ACUTE CORONARY SYNDROME
Multiple parameters of neutrophil activity in ACS patients have been studied, including peripheral neutrophil/lymphocyte ratio, neutrophil-specific microparticles, neutrophilic granular protein (neutrophil elastase, MPO, and MMP) release, neutrophil extracellular traps (NET) release, and interactions with tissue factor (Figure 1 and Table 1).
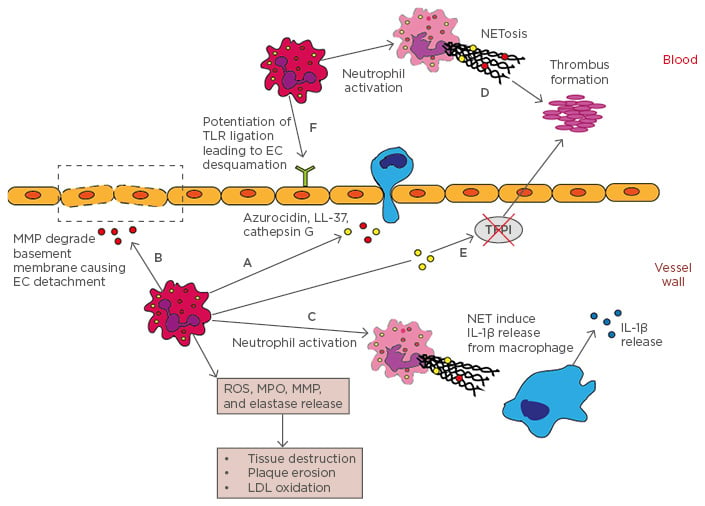
Figure 1: Neutrophils in atherosclerosis.
A) Neutrophils release azurocidin, LL-37, cathepsin G, MPO, and MCP-1, which are monocyte chemoattractants; B) Neutrophilic MMP degrade basement membrane causing endothelial cell detachment; C) NET complexes stimulate proinflammatory cytokine, IL-1β, release from macrophages; D) Activated neutrophils release NET complexes that facilitate thrombus formation; E) Neutrophil elastase and cathepsin G inhibit tissue factor plasminogen inhibitor, leading to thrombus formation; F) Neutrophils potentiate TLR ligation leading to endothelial cell detachment and plaque erosion.
EC: endothelial cells; IL-1β: interleukin-1β; LDL: low-density lipoprotein; MCP-1: monocyte chemotactic protein-1; MMP: matrix metalloproteinases; MPO: myeloperoxidase; NET: neutrophil extracellular traps; ROS: reactive oxygen species; TFPI: tissue factor plasminogen inhibitor; TLR: toll-like receptor.
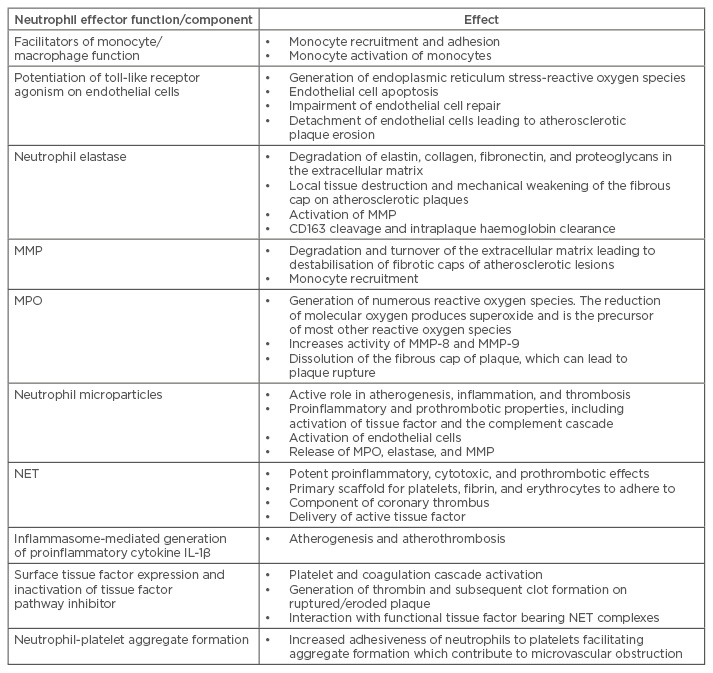
Table 1: Parameters of neutrophil function in acute coronary syndrome.
IL-1β: interleukin-1β; MMP: matrix metalloproteinases; MPO: myeloperoxidase; NET: neutrophil extracellular traps.
Peripheral white blood cell counts have been shown to be an independent prognostic factor in patients with ACS and positively correlate with the burden of coronary artery disease.31 The neutrophil to lymphocyte ratio is able to predict short and long-term mortality in patients with ACS and has correlated well with GRACE and SYNTAX scores, which are common clinical scoring systems that predict outcomes in ACS. Systemic polymorphonuclear counts have also been shown to be among the most robust predictors of acute coronary events and to directly impact short and long-term clinical outcomes.32 The low cost of measuring peripheral blood white blood cell counts is an additional advantage, facilitating its inclusion as an additional biomarker to predict outcomes in these patients.
Microparticles are small vesicles that arise from the plasma membrane of cells, and are commonly released after activation or early apoptosis. The microparticles play an active role in atherogenesis, inflammation, and thrombosis with circulating leukocyte-derived microparticles having been shown to be predictive of atherosclerosis.33 Activated circulating neutrophils shed CD66b+ microparticles, which have proinflammatory and prothrombotic properties, including activation of tissue factor and the complement cascade.34 Neutrophil specific CD66b+ microparticles also activate endothelial cells and promote the release of proteolytic enzymes, such as MPO, elastase, and MMP-9, all of which can lead to local tissue destruction and mechanical weakening of the fibrous cap on atherosclerotic plaques. Sampling of the coronary sinus directly after percutaneous coronary intervention (PCI) has shown an increased release of neutrophil-derived CD66b+ microparticles and MPO in patients with ACS compared to stable angina.35 These findings suggest that neutrophils are activated in vulnerable atherosclerotic plaques, with CD66b+ microparticles either being embedded within the coronary plaque/thrombus complex and released by mechanical disruption from PCI, or de novo activation of neutrophils as a result of PCI exposing neutrophils to cholesterol crystals (which are more abundant in vulnerable plaques compared to stable plaques), leading to cell death and the release of NET.36
Neutrophil elastase is a serine protease that causes degradation of elastin, collagen, fibronectin, and proteoglycans in the extracellular matrix. Neutrophil elastase has been detected in the shoulder region of atherosclerotic plaques, suggesting that it might play a role in the remodelling of atherosclerotic plaques from its direct effects on extracellular matrix degradation, as well as activation of MMP.37 Neutrophil elastase also cleaves CD163, a monocyte/macrophage cell surface receptor for haemoglobin-haptoglobin complexes, which clears cell free haemoglobin generated from haemolysis of extravasated red blood cells. The clearance of haemoglobin (which can deposit within atherosclerotic plaques as a result of intraplaque haemorrhage) is significantly impaired by neutrophil elastase induced CD163 cleavage.23 The resultant accumulation of plaque haemoglobin promotes inflammation and oxidative stress, and can then lead to plaque instability and vulnerability. ACS patients have been shown to have higher neutrophil elastase levels compared to stable angina and control patients, independent of traditional cardiovascular risk factors.38
Another important group of neutrophilic enzymes are the MMP. MMP-1, 8, 9, and 13 are responsible for the degradation and turnover of the extracellular matrix and can lead to destabilisation of fibrotic caps of atherosclerotic lesions, causing plaque rupture. These MMP have been shown to accelerate atherogenesis and extracellular matrix degradation in both animal and human studies.39 Clinical studies of ACS patients have also shown that MMP levels correlate with optical coherence tomographyproven plaque rupture and are predictive of future cardiovascular events.40,41
Activated neutrophils also produce MPO, a member of the haem peroxidase superfamily, which is stored in the azurophilic granules of the neutrophil and is responsible for the generation of numerous ROS. The reduction of molecular oxygen (O2) produces superoxide (O2•-) which is the precursor of most other ROS.42
Investigation of MPO in animal models of atherosclerosis have produced variable and contrasting results.43 In contrast to animal studies, the presence of MPO in human atherosclerosis has been consistently found. ROS have been associated with multiple cardiovascular adverse events, including atherosclerosis, heart failure, and hypertension.44 MPO typically uses hydrogen peroxide to generate hypochlorous acid, which has microbicidal properties but also disrupts normal vascular haemostasis. MPO-generated ROS increase the activity of MMP-8 and MMP-9.45 Serum MPO levels have been shown to correlate with severity of angiographic coronary artery disease,46 as well as being predictive of subsequent cardiac events (nonfatal myocardial infarction, death, and need for coronary revascularisation) in patients presenting with either chest pain or ACS.47 Elevated levels of MPO have also been described at sites of plaque erosions in patients with ACS,48 postulating that activated neutrophils adhere to atherosclerotic plaques with impaired endothelial integrity due to erosive events. In addition to this, MPO has been shown to also cause dissolution of the fibrous cap of a plaques, which leads to plaque rupture and superimposed thrombus formation.49
Another neutrophil function that has been gathering interest is their ability to release NET. The release process of these chromatin complexes from the nucleus of neutrophils is referred to as NETosis, which is a cell death programme different to apoptosis or necrosis and helps neutrophils to more efficiently eliminate pathogens by forming a mechanical barrier to ‘trap’ pathogens.50 NET are made up of neutrophilic granular and cytosolic proteins, proteases, and chromatin, which are wound in sequence around histone protein cores.51 Citrullination of histones via peptidylarginine deiminases (PAD) is essential in NETosis.50 Recent evidence has emerged suggesting that in addition to infectious disease, NET also play a role in non-infectious disease processes, including atherosclerosis and thrombosis,52 and this has resulted in a plethora of studies looking at the specific effects of NET in modulating components of the immune system causing atherogenesis. The release of NET causes potent proinflammatory, cytotoxic, and prothrombotic effects53,54 and serves as a primary scaffold for platelets, fibrin, and erythrocytes to adhere to, thus forming a link between inflammation and thrombosis. Intracoronary thrombus aspirate samples from patients with acute myocardial infarction revealed that high mobility group box 1 proteins, which were presented to neutrophils by activated platelets, induced the formation of NET.55 This highlights the interaction between neutrophils and platelets in atherothrombosis and postulates that NET could contribute to plaque rupture. The quantity of NET formation in coronary artery thrombus in patients with STEMI has also been shown to correlate positively with enzymatic and magnetic resonance imaging (MRI) measured infarct size and negatively with ST segment resolution.56 One suggested theory is that coronary NET may be propagating thrombosis and inflammation distally into the area of infarction causing further myocyte necrosis, leading to dysfunction of the microvasculature.57 Another study of thrombus aspirates in STEMI patients undergoing PCI revealed neutrophils release NET which, in turn, expose active tissue factor in infarct-related areas, but not in non-infarcted regions.58 This suggests that local release of NET in sites of plaque rupture contribute to atherothrombosis via the delivery of active tissue factor. Nucleosomes and double-stranded DNA, which are key components of NET, have been shown to be independently associated with severe coronary atherosclerosis59 and this has led to nucleosomes and double-stranded DNA being used as surrogate markers of NETosis.
Atherosclerotic plaque rupture exposes its prothrombotic contents (modified lipids, highly inflammatory cells, and necrotic core) to circulating blood, resulting in platelet and coagulation cascade activation, which ultimately leads to atherothrombosis via fibrin deposition and entrapment of blood cells.60 Activated neutrophils are major contributors to arterial thrombosis after plaque rupture. Circulating neutrophils express tissue factors on their cell surfaces and their activation can lead to the generation of thrombin and subsequent clot formation.61 As discussed earlier, neutrophils also release tissue factors through NET and functional tissue factor bearing NET have been identified at culprit artery sites of patients with STEMI.58 Activated neutrophils within atherosclerotic plaques can also inactivate tissue factor pathway inhibitor, which is an important regulator of tissue factor-induced blood coagulation, via the release of proteases at the site of vessel injury.62 ROS generated by activated neutrophils within atherosclerotic plaques also cause platelet activation. This accentuation of prothrombic processes by neutrophils can lead to significant clinical outcomes in ACS.
In particular, activated circulating neutrophils promote microvascular obstruction via neutrophil plugging and microthrombi formation in the microcirculation, leading to increased infarct size.63 The proinflammatory state in ACS leads to increased neutrophil-platelet adhesion with CRP and IL-6 levels strongly correlating to neutrophilplatelet aggregate formation detected by flow cytometry.64 Moreover, an abundance of highly activated neutrophil/platelet aggregates has been identified in aspirated coronary thrombus in STEMI patients undergoing primary PCI.56 These neutrophils express adhesion markers such as CD11a, CD11b, and CD66b, all of which increase adhesiveness of neutrophils to platelets, facilitating aggregate formation that can further contribute to microvascular obstruction.
POTENTIAL THERAPEUTIC OPTIONS
As the role of neutrophils in cardiovascular disease is increasingly appreciated, potential treatment strategies targeting NET release and breakdown have been studied. Deoxyribonuclease (DNAse), an enzyme that breaks down NET, has been shown in a clinical study of STEMI patients to accelerate lysis of coronary thrombi.56 The study demonstrated that DNAse levels correlated negatively with coronary NET burden and cardiac MRI-measured infarct size. DNAse treatment in a murine model of myocardial ischaemia resulted in decreased infarct sizes and infiltration of neutrophils in infarcted myocardium, as well as reduced circulating nucleosome levels, an indirect measure of NETosis.65 Another therapeutic agent targeting NET are PAD inhibitors. PAD are enzymes involved in citrullination of histones and the use of Cl-aminidine, a peptidomimetic pan-PAD inhibitor, in apolipoprotein E-deficient mice resulted in significantly reduced atherosclerotic lesion areas.66 Colchicine, an anti-inflammatory agent, has been shown to acutely suppress caspase-1 activity and inhibit inflammasome activation in monocytes from ACS patients. Similarly, targeting the neutrophil inflammasome or inhibiting its assembly could also be a therapeutic strategy in reducing the athero-inflammatory cytokine IL-Iβ.11,13
In summary, there are abundant data demonstrating the role of neutrophils in ACS. This knowledge presents opportunities to develop novel diagnostic and prognostic biomarkers and therapeutic agents that specifically target neutrophil activation and/or downstream mediators.