Abstract
Cardiac diseases have complex molecular origins. However, current clinical diagnostic tools are often inadequate to uncover specific molecular components of cardiac pathologies. Thus, we are still lacking a detailed understanding of disease progression, and both patient diagnosis and treatment are often inaccurate. Molecular imaging could play a leading role in translating basic research to both preclinical and clinical cardiac research, ultimately improving our understanding and management of human disease. In this review, we highlight the diversity of current molecular imaging tools that have been used in clinical research or have reached the stage of clinical translation. Facilitated by the steadily increasing infrastructure of clinical positron emission tomography and positron emission tomography-magnetic resonance imaging cameras and advancing gating analysis, these tools allow the implementation of clinical cardiac molecular imaging trials to deepen our knowledge of human disease and improve patient care.
INTRODUCTION
Cardiovascular disease remains the leading cause of death in Europe and worldwide.1 Almost all major pharmaceutical companies run large programmes on cardiovascular and cardiac drug development. Molecular imaging may play an important role in improving not only diagnosis but also treatment of cardiac diseases. The current understanding of molecular heart function is sufficient to implement more molecular imaging tools in the clinics for interrogating major aspects of cardiac function, including metabolism, innervation, and conduction. The infrastructure for clinical positron emission tomography (PET) and PET-magnetic resonance imaging is steadily growing, allowing for routine application of cardiac scans, including motion correction for heartbeat and breathing. The purpose of this review is to provide an overview of established and upcoming molecular imaging probes for cardiac function, which could aid our understanding of the human heart in health, disease, and as a function of treatment.
METABOLISM
In contrast to skeletal muscle, cardiomyocytes sustain an everlasting cycle of contraction and relaxation in order to feed our body with blood and maintain the homeostasis of nutrients and metabolic gases.2 This highly specialised task is facilitated by a specific molecular architecture. With their steady and sustained workload, cardiomyocytes are specialised for aerobic metabolism of fatty acids (FA) and are packed with mitochondria performing oxidative phosphorylation and β-oxidation. This unique molecular architecture offers numerous opportunities to use cardiac FA metabolism as a platform for molecular imaging (Figure 1). Although cardiac metabolism has a wide adaptive capacity and plasticity when facing challenging heart energy production conditions, most forms of cardiac diseases are associated with acute or chronic changes in energy metabolism.
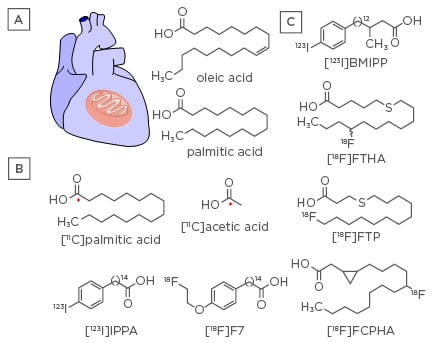
Figure 1: Cardiac fatty acid metabolism.
A) Chemical structures of native unsaturated and saturated FA, oleic acid, and palmitic acid. B) PET and SPECT radiotracers that are isotopes of native FA ([11C]palmitic acid and [11C]acetic acid), or mimic their metabolism ([123I]IPPA and [18F]F7). C) FA analogues as SPECT ([123I]BMIPP) and PET ligands ([18F]FTHA, [18F]FTP, and [18F]FCPHA) that facilitate mitochondrial trapping.
BMIPP: β-methyl-para-[123I]-iodophenyl-pentadecanoic acid; F7: fluoroethoxy phenyl pentadecanoic acid; FA: fatty acid; FCPHA: fluoro-3,4-methyleneheptadecanoic acid; FTHA: fluoro-6-thia-heptadecanoic acid; FTP: fluoro-4-thia-palmitate; IPPA: pentadecanoic acid; PET: positron emission tomography; SPECT: single photon emission computed tomography.
The heart rapidly extracts FA via either passive diffusion or a protein carrier-mediated pathway.2,3 Oleate and palmitate are the most abundant FA in the blood pool. Once inside the cytosolic compartment of the cardiomyocyte, FA are esterified to long-chain acyl-coenzyme A (CoA) esters by FA-CoA synthase. The activated FA can then be esterified to triacylglycerols, or transferred to carnitine via carnitine palmitoyltransferase 1 (CPT1). The acylcarnitine is then shuttled into the mitochondria by carnitine-acylcarnitine translocase, where the majority is converted back to FA-CoA by carnitine palmitoyltransferase 2 and enters mitochondrial FA β-oxidation.
Ventricular hypertrophy and dilated cardiomyopathy (DCM) are characterised by decreased myocardial capacity of FA oxidation and a shift to glucose metabolism, as a result of a decreased expression and activity of proteins involved in FA metabolism.4,5 By contrast, diabetic cardiomyopathy results in an increase of FA oxidation due to increased circulation levels and consequential accumulation of intramyocardial lipid metabolites.2 Ischaemic heart disease involves reduced oxidative metabolism due to reduced oxygen supply and increased anaerobic glycolysis, even though FA oxidation recovers quickly during reperfusion following ischaemia.2 Given this molecular pathology, current PET imaging of myocardial metabolism utilises 11C-labelled natural FA and 18F-labelled FA analogues, as well as the canonical [18F]fludeoxyglucose.6 For the purpose of this review, we will focus on FA metabolism, which is an independent predictor of left ventricular mass in hypertension and left ventricular dysfunction.7
[11C]palmitate has been used to assess various steps of myocardial FA metabolism in the human heart, including storage as a triglyceride and β-oxidation (Figure 1B). Given the small storage capacities of cardiomyocytes for triglycerides and the vast number of mitochondria, the signal is dominated by the oxidation pathway and can thus be used for evaluating the enzymatic activity of CPT1, the enzyme that catalyses the acyl transfer from FA-CoA to carnitine.8 Using this imaging probe, it has been demonstrated that obesity and diabetes mellitus (DM) are associated with an increase in myocardial FA metabolism and reduced glucose utilisation.9,10 By contrast, patients with idiopathic DCM exhibited alterations in myocardial metabolism characterised by decreased FA oxidation and increased glucose metabolism.11 Thus far, the routine clinical use of [11C]palmitate has been limited by its relatively complicated synthesis and a need for an on-site cyclotron. Recent efforts to improve synthesis and a growing infrastructure of hospital cyclotrons may facilitate future clinical use of this radiotracer.12
Analogously to FA, acetate is rapidly extracted by cardiomyocytes and oxidised in mitochondria via the tricarboxylic acid (TCA) cycle to CO2 and H2O.13 [11C]acetate has been used to measure myocardial blood flow in patients with hypertrophic cardiomyopathy14 and enabled simultaneous quantification of myocardial perfusion, oxidative metabolism, cardiac efficiency, and pump function at rest as well as during exercise in athletes.15
15-(p-[123I]iodophenyl)-pentadecanoic acid ([123I]IPPA) was the first single photon emission computed tomography (SPECT) ligand to image myocardial FA metabolism in vivo, showing rapid accumulation in the heart and similar clearance kinetics as palmitate, which are directly correlated with β-oxidation in animal disease models and in humans.16,17 Currently, [123I]IPPA is commercially available in Europe and Canada, and its usefulness as a cardiac imaging agent has been explored in clinical trials.
Tu et al.18 designed an 18F-labelled radiotracer, 15-(4-(2-[18F]fluoroethoxy)phenyl)pentadecanoic acid ([18F]F7), based on [123I]IPPA. [18F]F7 is capable of mimicking [11C]palmitate with regard to its ability to capture all key aspects of FA metabolism, including uptake, β-oxidation, and storage as a triglyceride. In addition, it is more feasible for clinical use due to the longer half-life of 18F, and it allows quantitative PET imaging with improved temporal resolution compared to SPECT. Small animal PET studies in Sprague-Dawley rats displayed high uptake in the heart, good imaging contrast over blood and other tissues, and similar biphasic washout kinetics as [11C]palmitate.
In addition to isotopes of native FA, FA analogues acting as false substrates or inhibitors of FA metabolism have been explored as PET and SPECT ligands, whose signal emphasises myocardial β-oxidation (Figure 1C). These analogues are recognised by the cytoplasmic acyl-CoA synthase and CPT1 in the mitochondrial membrane but are trapped inside the mitochondria due to incomplete β-oxidation.
For example, β-methyl-p-[123I]-iodophenylpentadecanoic acid ([123I]BMIPP) has been developed as a derivative of the aforementioned [123I]IPPA with the intention of preventing β-oxidation by blocking the β-position with a methyl functional group, thus enforcing accumulation in mitochondria. [123I]BMIPP imaging has been used for identifying patients with recent exerciseinduced myocardial ischaemia19 and non-invasive diagnosis of coronary artery disease in patients with heart failure.20
Pandey et al.21 investigated long-chain [18F] fluorothia FA for their use as myocardial β-oxidation probes. CoA thioesters of 4-thia FA analogues are potent inhibitors of β-oxidation through inhibition of acyl-CoA dehydrogenase, thus leading to an accumulation of radiotracers in the mitochondria after incomplete β-oxidation.22-24
14(R,S)-[18F]Fluoro-6-thia-heptadecanoic acid ([18F]FTHA) uptake was reduced by 81% in mice after pharmacological inhibition of CPT1 using 2-[5-(4-chlorophenyl)pentyl]-2oxiranecarboxylate.25 Likewise, β-oxidation inhibition by lactate infusion in pigs reduced the myocardial [18F]FTHA signal by 89%, demonstrating that nearly all of the PET ligand taken up by the heart enters the mitochondria.26,27 Both studies confirmed that the unidirectional uptake rate of [18F]FTHA reflects β-oxidation and allows the measurement of cardiac FA-metabolism in vivo, which has been exploited in numerous clinical studies. For example, accumulation of [18F]FTHA in the myocardium was increased by aerobic exercise in human volunteers due to increased energy demand.28 In patients with coronary artery disease, [18F]FTHA imaging displayed a reduced signal in accordance with reduced oxidative metabolism.29 In addition, the [18F]FTHA signal was reduced in healthy volunteers with glucose/insulin clamp, demonstrating the ability to detect a change in energy substrate preference from FA to glucose.30 Furthermore, [18F]FTHA has recently been used to investigate the role of metabolic alterations in the development of a maladaptive right ventricular response in pulmonary arterial hypertension.31
Following the same chemical strategy of inhibiting β-oxidation, 16-[18F]fluoro-4-thia-palmitate ([18F] FTP) was developed to improve sensitivity to changes in FA oxidation caused by hypoxia, and was found to have prolonged myocardial retention.32 [18F]FTP showed reduced FA oxidation in hypoxic rat hearts, showing comparable imaging qualities as [18F]FTHA in swine, but was not influenced by altered plasma substrate levels.33 Elevated uptake of [18F]FTP was found in patients with Type 2 DM.34 This is in line with the aforementioned increase in myocardial FA oxidation found in this patient group using [11C]palmitate.
Shoup et al.35 introduced trans-9-[18F]fluoro-3,4methyleneheptadecanoic acid ([18F]FCPHA), which features a cyclopropyl group to block β-oxidation, as a new radiolabelled FA analogue. [18F]FCPHA revealed fast blood clearance, high myocardial uptake, and good retention in rats and rhesus monkeys. [18F]FCPHA is currently undergoing a Phase II clinical trial to assess myocardial FA perfusion and uptake in coronary artery disease patients.36
INNERVATION
Heart function is controlled by the autonomous nervous system (Figure 2). While cholinergic neurons dominate innervation to the sinoatrial node, setting a constant rhythm of contractions,37 adrenergic fibres predominantly innervate the ventricles.38 Adrenergic activity induces several cardiovascular effects, including an increase in cardiac contractility (inotropy), frequency (chronotropy), rate of relaxation (lusitropy), and acceleration of impulse conduction through the atrioventricular node (dromotropy).39 These effects are triggered by the catecholamine neurotransmitters noradrenaline and adrenaline acting on β1-adrenergic receptors.38
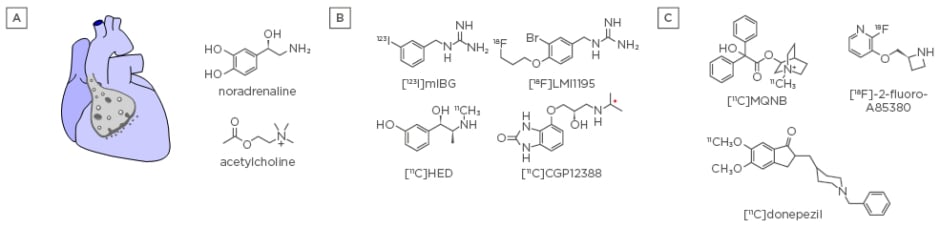
Figure 2: Cardiac innervation.
A) The heart is innervated by sympathetic and parasympathetic neurons, which signal through the neurotransmitters noradrenaline and acetylcholine, respectively. B) SPECT and PET radiotracers for assessing adrenergic innervation by measuring the presynaptic NAT ([123I]mIBG, [11C]HED, and [18F] LMI1195) and β1-receptors ([11C]CGP12388). C) PET radiotracers for assessing parasympathetic innervation of the heart by measuring muscarinic receptors ([11C]MQNB), nicotinic α4β2 receptors ([18F]-2-fluoro-A85380), and AChE ([11C]donepezil). AChE: acetylcholinesterase; mIBG: meta-[123I]iodobenzylguanidine; MQNB: N-[11C]methyl-quinuclidin-3-yl benzilate; NAT: noradrenaline transporter; PET: positron emission tomography; SPECT: single photon emission computed tomography.
The importance of the sympathetic nervous system in cardiac disorders, including ischaemic heart disease and heart failure, is well established.40 Aberrant adrenergic function, including increased neurotransmitter release and reduced reuptake, occurs early in the development of heart failure as a compensatory mechanism for sustaining the cardiac output despite, for instance, a physical insult to the myocardium or increased peripheral resistance. However, if increased activity persists over time, the heart will progress into a state of chronic decompensated heart failure, and the hyperactive adrenergic activity will continue to stimulate the heart to work at a level much higher than the cardiac muscle can handle.38
This pathological adrenergic innervation can be measured using molecular imaging, for example SPECT and PET. Even though the cardiac specific isoforms of proteins involved in adrenergic signalling would allow, in theory, the development of dedicated and cardiac-specific imaging tools, the current toolbox is centred on general mimetics of the native neurotransmitters (Figure 2B).
Meta-[123I]iodobenzylguanidine ([123I]mIBG), an iodinated adrenergic neurotransmitter analogue, is commonly used for SPECT imaging of the presynaptic noradrenaline transporter (NAT).41 NAT is a transmembrane protein that functions as a rapid noradrenaline reuptake system located at or near presynaptic terminals, and terminates noradrenergic signaling.42 The cardiac signal of [123I]mIBG is lower in individuals with heart failure due to a higher NAT occupancy, allowing the use of [123I]mIBG as an independent predictor of heart failure progression and cardiac mortality. [123I]mIBG has been used clinically in different continents for almost three decades.43-45
In order to make adrenergic innervation imaging available for quantitative and non-invasive PET imaging, 11C and 18F-labelled ligands have been developed (Figure 2B). For example, [11C]metahydroxyephedrine ([11C]HED) was developed based on metaraminol,46 a synthetic false transmitter analogue of noradrenaline, which is transported by NAT. It is, however, resistant to catechol-O-methyl transferase and monoamine oxidase metabolism and is thus retained in nerve terminals due to continuous cyclical release and reuptake.47 [11C]HED displayed a lower myocardial signal in patients with congestive heart failure,48 which correlated to patient prognosis.49 The 18F-labelled N-[3-bromo-4- (3-[18F]-fluoro-propoxy)-benzyl-guanidine ([18F] LMI1195) is structurally related to [123I]mIBG and showed a reduced signal in rodent models of heart failure.50 Additionally, first-in-human clinical trials showed its potential for assessing myocardial sympathetic activity in vivo.51 The sensitive and quantitative PET imaging using [11C]HED and [18F]LMI1195 allowed evaluation of regional denervation and heterogeneity of innervation in the heart, and could be used for predicting sudden cardiac death.48,51
Following the chemical design of [123I]mIBG, 18F-labelled benzylguanidines (meta-[18F] fluorobenzylguanidine ([18F]mFBG)52 and para-[18F]fluorobenzylguanidine ([18F]pFBG)) have been developed as alternative probes for imaging NAT using PET.53,54 Both candidates have been successfully tested in neuroendocrine tumour mouse models but not yet for sympathetic nervous system dysfunction in cardiovascular diseases.
As a consequence of sustained enhanced sympathetic stimulation, postsynaptic β-adrenoceptors are downregulated in heart failure.55,56 The recently disclosed 11C-labelled beta-blocker S-4-(3-([11C]-isopropylamino)-2hydroxypropoxy)-2H-benzimidazol-2-one ([11C] CGP12388) (Figure 2B) allowed measurements of postsynaptic β1-receptor density and occupancy in patients with idiopathic DCM.57 Thus, the current molecular imaging toolbox for sympathetic innervation enables measurements of all major pre and postsynaptic components of cardiac adrenergic signalling in humans. Given the widespread use of beta-blockers, it will be interesting to interrogate receptor dynamics as a function of (long-term) treatment.
Abnormal function of the parasympathetic system, in particular parasympathetic withdrawal, has been recognised as a key component of the molecular pathology underlying heart disease.58,59 The highly specific muscarinic acetylcholinergic antagonist N-[11C]methyl-quinuclidin-3-yl benzilate ([11C] MQNB) has been used to measure myocardial muscarinic acetylcholine receptor (mAChR) density in vivo (Figure 2C).60 [11C]MQNB measurements in idiopathic DCM patients revealed an upregulation of myocardial mAChR, presumably as an adaptive mechanism to β-adrenergic stimulation.61 Likewise, patients with myocardial infarction showed increased [11C]MQNB signal in non-damaged left ventricular regions.62 Since there is a decreased vagal tone in these pathologies, the role of upregulated expression of mAChR has still to be elucidated, and molecular imaging could play an integral role in understanding the dynamics between sympathetic and parasympathetic signalling in humans.
Acetylcholinesterase (AChE) inhibitors, such as pyridostigmine, have been used as parasympathomimetica, for instance in chronic heart failure patients.63 Gjerløff et al.64 tested [11C]donepezil for measurements of AChE density in human peripheral organs and found a homogenous and displaceable signal of the tracer in the left ventricle of the heart, which might help in understanding the role of neurotransmitter metabolism in human disease (Figure 2C).
Imaging of nicotinic AChR could help in better understanding their pathophysiological role in atherosclerotic disease progression and could thus be used as a diagnostic tool to risk-stratify patients for myocardial infarction and stroke.65,66 [18F]-2-fluoro-A85380 was the first PET radiotracer developed for imaging of the cardiac α4β2-nicotinic AChR in healthy subjects and has already been tested in patients with Parkinson’s disease and patients with multiple system atrophy (Figure 2C).67
Given the currently available imaging technology for muscarinic and nicotinic receptors, as well as AChE, understanding the dynamics of different components of the cholinergic innervation system in humans is within reach and would provide major scientific and clinical impact at relatively low risk of failure.
MYOCARDIAL CONDUCTION
The neuronal input from sympathetic and parasympathetic transmission is translated into cardiac action through a series of ion channels that respond to changes in membrane potential and ultimately couple excitation to contraction (EC-coupling).68
Fast activating voltage-gated sodium channels (NaVs), mainly NaV1.5 (SCN5A), initiate action potentials and strongly depolarise cardiomyocytes (Figure 3).69 Subsequent activation of high-voltage activated Ca2+ channels (L-type, mainly CaV1.2) leads to Ca2+ influx and further Ca2+ release from the sarcoplasmic reticulum, which increases the cytosolic Ca2+ concentration from nanomolar to micromolar concentrations.70 This increase elicits conformational changes of the filament complex to facilitate the actin-myosin interaction and attains myocardial contraction.68
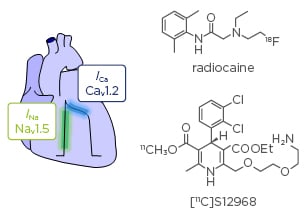
Figure 3: Cardiac conduction.
Electrical signalling through the myocardium couples excitation to contraction by the action of voltage-gated ion channels. Radiocaine imaging allows quantifying cardiac voltage-gated sodium channels in vivo using PET. The dihydropyridine derivative [11C]S12968 enables measurements of L-type calcium channels in the heart.
PET: positron emission tomography.
Numerous cardiac diseases are rooted in abnormal electrical signalling of the myocardium due to mutated ion channels or pathological expression levels.71 Heterozygous mutations in SCN5A have been implicated in rare genetic arrhythmia, such as Brugada syndrome, long-QT syndrome, and progressive cardiac conduction defect. Abnormal expression levels of SCN5A have also been found in structural heart disease, including heart failure and ischaemic cardiomyopathy.72 Furthermore, an increase in dihydropyridine (DHP) receptor expression, an L-type calcium channel, has been found in hypertrophied hearts.73
Recently, the first PET radiotracer for in vivo molecular imaging of cardiac SCN5A has been developed (Figure 3).74 Radiocaine, an analogue of the classical Class-1b antiarrhythmic lidocaine, allows in vivo measurements of density and drug occupancy of cardiac SCN5A, which, to date, could only be determined invasively or through in vitro methods. Specific binding to SCN5A has been demonstrated in living rats and baboons; furthermore, autoradiography studies using myocardial tissue from human failing heart explants revealed reduced target density.
In order to assess changes in L-type calcium channel density in vivo, several DHP-based drugs have been radiolabelled with 11C for PET imaging (Figure 3).75 The amlodipine analogue [11C]S12968 showed ≤80% specific binding in the myocardium and was used for in vivo measurement of myocardial DHP binding site density in beagles, with low doses of Ca2+ channel antagonists.
The ability to measure both NaVs and CaVs as determining components of the EC-coupling opens the door for future clinical studies, which will precisely identify the molecular dynamics of myocardial ion channel signalling in human disease.
PERSPECTIVE
Taken together, there is a wealth of wellunderstood tools for in vivo molecular imaging of cardiac function and health, including oxidative metabolism, adrenergic and cholinergic innervation, and myocardial electrical conduction (Table 1). The majority of these tools have already been validated in humans, displaying minor radiation exposure, or have reached the stage of clinical translation, allowing widespread clinical studies in the future. Further evaluation in clinical trials will provide data on the impact of molecular imaging on changing diagnosis, treatment, and prognosis. We are convinced that the growing infrastructure of clinical PET magnetic resonance cameras will play an integral role in expanding the use of molecular imaging in cardiology, in particular with recent advances in cardiac gating reconstruction.76 The combined information of structural integrity and changes in the cardiac molecular machinery will not only yield impactful insights in basic research but will also improve individualised patient care.
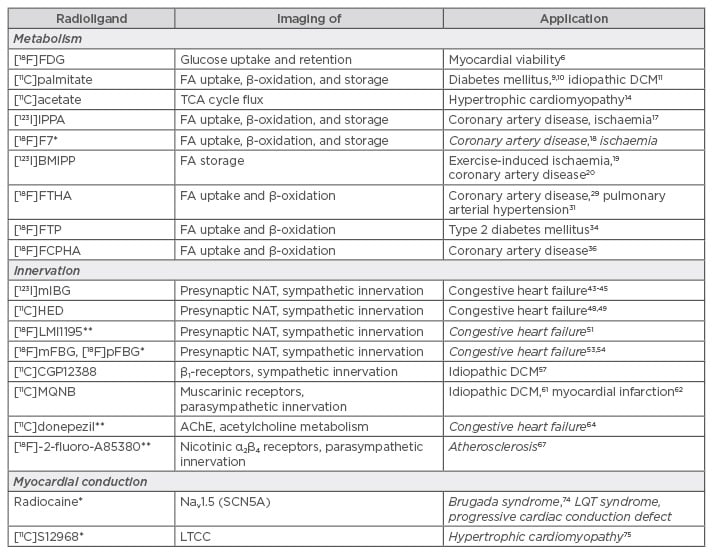
Table 1: Summary of molecular imaging tools and their (bio)medical application(s).
*Only tested in animal models; **only tested in first-in-human clinical trials; potential future applications in italics.
AChE: acetylcholinesterase; DCM: dilated cardiomyopathy; FA: fatty acid; LTCC: L-type calcium channel; Nav: voltage-gated sodium channels; NAT: noradrenaline transporter; TCA: tricarboxylic acid; LQT: long-QT.