Abstract
A growing body of evidence describes a strict relationship between bone and cardiovascular disease (CVD), with several physiological and pharmacological implications. Advances in our understanding of the pathophysiology of osteoporosis and vascular calcifications indicate that these two processes share common pathogenetic mechanisms, suggesting the existence of a bone–vascular axis. Indeed, the hypothesis that calcium deposition in arteries may be an active process of extracellular matrix mineralisation and of ectopic ossification is advancing. In this regard, molecular markers of osteogenic activity can be found in all arterial calcified segments. Nevertheless, the panel of bone-related markers of CVD is still unclear. This review focusses on molecules serving well-defined roles as bone markers (namely osteocalcin, osteoprotegerin, sclerostin, osteopontin, and klotho) and on which possible novel roles may exist as markers of CVD. A detailed understanding of novel bone-related markers of CVD is necessary to address the unmet clinical needs of an increasingly aged and dysmetabolic population.
INTRODUCTION
Vascular calcification is an independent risk factor for cardiovascular disease (CVD). Calcium deposits in coronary arteries can weaken vasomotor responses and alter the stability of atherosclerotic plaques, increasing the risk of cardiovascular events. Vascular calcification has historically been considered as a passive process caused by cellular death; recently however, the hypothesis that calcium deposition in arteries may be an active process of extracellular matrix mineralisation is gaining traction. In this regard, molecular markers of osteogenic activity can be found in all arterial calcified segments.1 Interestingly, a strict relationship between vascular and bone diseases has been described. Advances in our understanding of the pathophysiology of osteoporosis and vascular calcifications indicate that these two processes share common pathogenetic mechanisms, suggesting the existence of a bone–vascular axis.² Factors implicated in the pathogenesis of both osteoporosis and vascular calcification include proteins, hormones, lipids, vitamins, and cellular activities.3
Bone morphogenetic proteins participate in osteoblast differentiation while simultaneously producing reactive oxygen species and increasing the adhesiveness of monocytes on the vascular wall. In this review, the authors describe the epidemiological evidence and mechanisms underlying vascular calcification compared to bone calcification, and discuss in detail biomarkers implicated in the bone–vascular axis.
THE BONE-VASCULAR AXIS
Epidemiological Evidence
Several epidemiological studies have demonstrated an association between CVD and bone diseases, including osteoporosis. Both osteoporosis and CVD are age-related disorders, and the temporal link between osteoporosis and vascular calcification is particularly marked in post-menopausal women and the elderly.4 During menopause, progression of aortic calcification is associated with metacarpal bone loss.⁵ In post-menopausal women, osteoporosis is strongly associated with the presence of breast arterial calcification and increased severity of carotid atherosclerosis, with impaired brachial arterial endothelial function, increased arterial stiffness, and the presence of coronary atherosclerosis.⁶
Rodriguez et al.⁵ demonstrated that lower bone mass density (BMD) was marginally, but significantly, correlated with increased cardiac workload (measured as the rate pressure product). This relationship was partially mediated by abdominal aortic calcification, suggesting that elderly with osteoporosis, particularly females, may have an increased cardiovascular risk. The Framingham Heart Study has also contributed to our knowledge in unveiling the existence of a bone–vascular axis, showing that loss in cortical bone is associated with long-term progression of atherosclerotic disease, with a doubled risk of hip fracture observed in those affected by CVD. This evidence has also been confirmed by several other studies showing that low BMD is associated with extensive arterial artery calcification and with atherosclerotic plaque burden.⁷ Tankò et al.⁴ showed that BMD at the proximal femur was associated with the severity of aortic calcification independent of age (r=-0.12–17.00; p<0.001).
Many studies have also demonstrated increased bone fragility among people with increased risk for CVD, including those with diabetes.⁸ An increased risk of osteoporotic fractures in both Type 1 and Type 2 diabetes mellitus patients has been largely documented,9-11 with some studies showing an association between the presence of diabetic cardiovascular complications and lower BMD.12 In this regard, many studies have also reported variation of serum levels of osteokines in blood samples from patients with diabetes.5,13,14 Nevertheless, it has been recently shown that circulating undercarboxylated osteocalcin (OCN) serum levels are independently associated with cardiovascular risk, determined by Z-score, in patients affected by metabolic syndrome, regardless of the presence of Type 2 diabetes mellitus.15
Mechanisms of Vascular Calcification Compared to Bone Calcification
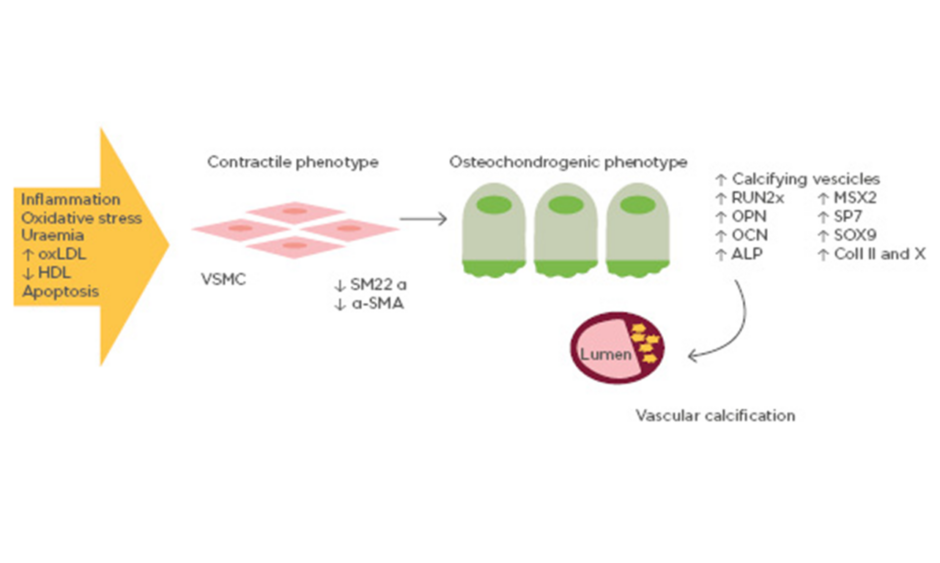
Pro-calcifying conditions, e.g., inflammation, oxidative stress, uraemia, increased oxidised low-density lipoprotein (oxLDL), decreased high-density lipoprotein, and apoptosis trigger the cellular reprogramming and phenotype switching of vascular smooth muscle cells (SMC) from a contractile to bone-forming state. Reprogrammed vascular SMC express bone related proteins such as RUNX2 that act in a paracrine way on all vessel pluripotent mesenchymal cells with concomitant downregulation of SMC contractile proteins, generate mineralised matrix vesicles which initiate the mineralisation process, and form bone matrix within the vessel wall.
Vascular calcification is caused by the deposition of hydroxyapatite crystals in the arterial wall, both in the tunica intima and tunica media. Arterial intimal calcification is associated with the development of atherosclerotic plaques. Intimal calcification is characterised by microcalcification deposits within the fibrous caps of the atherosclerotic plaque, weakening the structure of the arterial wall and increasing the risk of plaque rupture.16 Microcalcifications originate from the apoptotic SMC or mineralising vesicles that are released by these cells.17 Arterial medial calcification (Monckeberg’s medial sclerosis) is a concentric process distinguished by macrocalcification, medial fibrosis, and arterial stiffness. This occurs in the absence of lipid accumulation and inflammatory cell infiltration, similar to intimal calcification.1
Although a variety of mechanisms have been proposed for vascular calcification, the trans-differentiation of vascular SMC from a contractile to an osteochondrogenic phenotype seems to play a key role. This phenotype is characterised by the loss of SMC markers (SM22 α and α-SMA) and the gain of osteochondrogenic markers (RUNX2, SP7, OPN, OCN, alkaline phosphatase, SOX9, Type II/X collagen), accompanied by the down-regulation of mineralisation inhibitory molecules and the production of a calcification matrix (Figure 1). The differentiation of vascular SMC towards an osteoblast-like phenotype has been confirmed in vitro and in vivo. In a transgenic mouse model, it has been demonstrated that vascular SMC in the tunica media are able to differentiate into chondrocytes and osteoblasts if exposed to oxLDL and reactive oxygen species which up-regulate the expression of the bone-related transcription factor RUNX2.18,19 RUNX2 induces osteoblastogenesis from mesenchymal stem cells, an essential pathway in the ossification process of the extracellular matrix. In particular, RUNX2 increases expression of the bone-related proteins OCN, sclerostin, and RANKL. Therefore, oxLDL in the arterial wall leads to arterial intimal calcification by vascular SMC differentiation into osteoblast-like cells, regulated by molecules that initiate and regulate osteoblastic and chondrocytic differentiation.
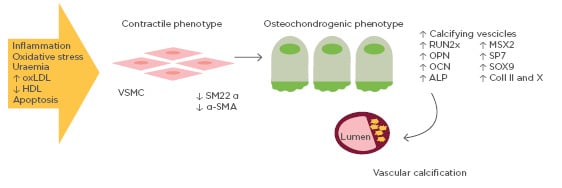
Figure 1: Mechanisms of vascular calcification involving bone-related proteins.
Vascular smooth muscle cells exposed to pro-calcifying conditions (e.g., inflammation, oxidative stress, high oxidised low-density lipoprotein) differentiate into osteoblast-like cells. Osteoblast-like vascular smooth muscle cells are able to express a series of osteoblast transcription factors, thus promoting vascular calcification.
ALP: alkaline phosphatase; Coll II and X: Type II and X collagen; HDL: high-density lipoprotein; OCN: osteocalcin; OPN: osteopontin; oxLDL: oxidised low-density lipoprotein; SM22 α: smooth muscle protein 22-α; α-SMA: smooth muscle α actin; RUNX2: runt-related transcription factor 2; VSMC: vascular smooth muscle cell.
OxLDL have also been shown to promote trans-differentiation of circulating monocytes towards a pro-calcific phenotype.20 Monocytes are well known for playing a key role in the development of atherosclerotic plaques through their transmigration to the arterial wall. Here, they differentiate into macrophages and contribute to the pro-inflammatory milieu, and phagocyte lipid droplets can give rise to foam cells. Recent evidence also strongly supports a role for monocytes in the active calcification process of arterial wall. Circulating osteoblastic progenitors were initially described in a landmark paper by Eghbali-Fatourechi et al.,21 in which mononuclear circulating OCN+ cells were able to form mineralised nodules when cultured in osteoblast-differentiating medium and cause ectopic calcification when transplanted in mice. Subsequently, monocytes have been described as a source of mesenchymal progenitors which can differentiate into osteoblast-like cells22 and contribute to atherosclerotic calcification.23 Some reports have suggested that circulating myeloid cells with osteogenic potential may affect CVD in the general population and in diabetes.21,24,25
Chronic inflammation seems to have a central role in the pathogenesis of medial calcification, which is often independent of atherosclerosis: this suggests different processes drive vascular SMC differentiation.26
Hyperglycaemia and dyslipidaemia cause the production of pro-inflammatory cytokines, such as TNFα primarily released by monocytes and macrophages. TNFα was found to be the main cytokine activating osteogenic programmes of vascular SMC via Msx2–Wnt signalling27 MSX2 is a gene coding for an osteochondrogenic transcription factor associated with intramembranous ossification. Wnt signalling is also implicated in osteoblast maturation, medial calcification, and fibrosis. Therefore, vascular SMC of the medial layer respond to different osteogenic stimuli, including inflammation or prolonged uraemia, and trans-differentiate into osteoblast-like cells, which can cause vessel wall stiffening through calcium deposits.1
BIOMARKERS OF THE BONE-VASCULAR AXIS i
Osteocalcin
OCN is the most expressed non-collagenous protein in the bone matrix, synthesised by osteoblasts. Several studies have shown an association between OCN and cardiovascular risk factors such as insulin resistance and dyslipidaemia.28 The relationship between OCN and atherosclerosis in humans has been suggested in different studies. In a study conducted in healthy post-menopausal women, there was an increased prevalence of carotid atherosclerosis in subjects with OCN levels above the median and low bone mineral density.29 Moreover, some evidence has shown that OCN is expressed in the calcific atherosclerotic lesions and in the vascular SMC of the vessel wall, suggesting a potential role in the differentiation of vascular SMC into osteogenic cells.30
Circulating osteoprogenitor cells, a member of the monocytic family, express OCN on their surface and exhibit increased abundance in patients with CVD.31 Because of their pro-calcific phenotype, these cells contribute to the development of vascular calcification and atherosclerosis under normal conditions.32 Patients with diabetes show elevated levels of OCN+ circulant monocytes compared to controls, especially in the presence of atherosclerotic CVD.31 Moreover, OCN+ cells have been found in coronaries of subjects with coronary artery disease, of which levels are associated with the presence of vascular calcifications and instability of the plaques.24
Osteoprotegerin
Osteoprotegerin (OPG) is a member of the TNF superfamily that acts by binding RANKL and TRAIL.33 Its role in bone metabolism is to inhibit osteoclastic bone resorption, a role it enacts through its presentation as a ‘decoy receptor’ for RANKL, preventing its binding to RANK and stimulating osteoclast activation. Indeed, elevated serum OPG levels are associated with higher BMD.34 Some evidence has shown that OPG could have a protective influence on the vascular system. OPG-knockout mice have concomitantly shown early onset osteoporosis and increased vascular calcifications.35 However, observational studies in humans suggest that OPG may be a marker of CVD. High levels of serum OPG have been associated with the presence of vascular calcifications,36 coronary artery disease,37 carotid atherosclerotic plaques,38 peripheral artery disease,39 and cardiovascular mortality.40
Furthermore, OPG also binds TRAIL, which is involved in the regulation and modulation of apoptosis.33 This binding also seems to play a direct role in the development of atherosclerotic plaques, with pleiotropic effects on the
vasculature. Some evidence has shown increased apoptosis to occur in TRAIL-treated endothelial cells, while other studies have described increased cellular survival and proliferation in response to TRAIL.41 Furthermore, it seems that high levels of TRAIL are expressed at the vulnerable plaque sites,33 and it is known that TRAIL stimulates the production of nitric oxide, which plays a protective role within the endothelium.42
Overall, the debate remains open as to whether increased OPG serum levels attenuate CVD or are instead representative of counter-regulatory protective pathway activation in the context of vascular calcification processes.
Sclerostin
Sclerostin is a soluble factor secreted by osteocytes. It regulates bone turnover by inhibiting Wnt/β-catenin signalling, which promotes the differentiation and proliferation of osteoblasts. Sclerostin is also able to stimulate osteoblast apoptosis through the activation of caspases.43 Therefore, when sclerostin action is suppressed, osteogenesis is indirectly stimulated.44 Loss of sclerostin gene function is related to diseases characterised by a hyperostosis process.43
Elevated serum sclerostin has also been associated with the presence of atherosclerosis and is a proposed marker of CVD, particularly in subjects with diabetes.45 Kuiper et al.46 showed that increased levels of sclerostin are associated with an augmented risk of having coronary artery calcifications. Sclerostin can also play a role in the development of valve calcification. Moreover, a study conducted by Leto et al.47 in 2018 showed that high levels of sclerostin were present in vascular smooth muscle cells of the atherosclerotic plaques in a cohort of patients who had undergone carotid endarterectomy, suggesting a potential role of this molecule in the development of atherosclerosis. Koos et al.48 showed that patients with aortic valve calcification have increased sclerostin levels compared to healthy controls, and that the severity of calcification is directly proportional with its serum levels.
Eventually, the presence of calcification in the abdominal aorta seems to be associated with elevated serum sclerostin levels, as emerged in a study conducted by Hampson et al.49 in 2013. This study showed that subjects with higher aortic pulse wave velocity, an indicator of arterial stiffness, had significantly higher serum sclerostin levels compared to subjects with normal pulse wave velocity.
Of note, it has been evidenced that the use of monoclonal anti-sclerostin antibodies for the treatment of post-menopausal osteoporosis can result in increased risk of cardiovascular adverse events,50 suggesting a possible protective role of sclerostin on the vasculature.
Osteopontin
OPN is a structural glycoprotein of the bone matrix. Because of its high number of aspartic acid residues, it can bind calcium and hydroxyapatite ions, inhibiting crystal formation. Additionally, it can bind several integrin receptors, especially integrin β3 on the surface of osteoclasts. This binding leads to a decrease in calcium concentration and to the activation of carbonic anhydrase II, both required for osteoclastic activation.2 These actions are important for the resorption of ectopic calcifications. OPN is also a regulator of calcification in the vessel wall. In a murine model, OPN has demonstrated to be an inhibitor of vascular calcification. In fact, Speer et al.51 showed that mice deficient in matrix Gla protein (a factor involved in the inhibition of bone mineralisation) and with deleterious OPN mutations are more prone to develop extensive vascular calcifications than mice deficient for matrix Gla protein alone. Studies conducted in humans confirmed that OPN could be a marker of vascular disease. The FinnDiane study showed that OPN is a strong predictor of cardiovascular events in subjects with Type 1 diabetes mellitus,52 whereas another study has shown an association between elevated OPN serum levels and the presence of coronary artery calcification in patients with Type 2 diabetes mellitus.53 Interestingly, some evidence has shown OPN to potentially be an inhibitor of vascular calcification, and may stimulate the dissolution of calcifications by inducing macrophages to express carbonic anhydrase.54 In this context, elevated serum levels of OPN may reflect a compensatory mechanism against calcification.
Klotho
Klotho is known as a membrane protein and as a circulating peptide (the extracellular domain is secreted into the blood), with peculiar anti-ageing properties. The membrane protein form is predominantly expressed in the distal tubule of the kidney, but also in multiple other tissues. In the kidney, it acts as a co-receptor for FGF-23, a bone-derived growth factor that mainly inhibits renal activation of vitamin D and induces tubular excretion of phosphate.55
Klotho is also supposed to be involved in the pathogenesis of arterial calcification.55 Mice with targeted deletion of klotho display severe osteoporosis and progressive atherosclerosis, as described in a study conducted by Kuro et al.56 in 1997. The relationship between levels of soluble klotho and the occurrence and severity of CVD has been shown in several clinical studies,57,58 and some polymorphisms of the gene have been related to the incidence of cardiovascular events.59
Lim et al.60 demonstrated for the first time that klotho and FGFR are expressed in human arteries, with downregulation in response to phosphorus and TNF. They also found that decreased klotho levels lead to increased calcification and demonstrated that upregulation of klotho (through vitamin D receptor activation by calcitriol) restored klotho/FGF-23 signalling, inhibiting vascular calcification.60
CONCLUSION AND FUTURE PROSPECTIVE
As widely demonstrated, vascular calcification is an independent risk factor for CVD and mortality, and the existence of a bone–vascular axis is emerging as a possible key pathway in the pathogenesis of CVD. Arterial calcification is not considered a passive process anymore, but it actively involves vascular smooth muscle cell reprogramming and phenotype switching to osteoblast-like cells attributable to pro-calcifying conditions, such as inflammation, oxidative stress, apoptosis, uraemia, and high serum calcium and phosphate levels. Nevertheless, an increasing number of bone-related biomarkers are being identified which may also contribute to the development of CVD (Table 1 and Figure 2). Future studies are needed to further understand the role of osteokines in the pathogenesis of vascular calcification and cardiovascular events,61 and to identify possible therapeutic targets to prevent arterial calcification reduction, and thereby, arterial stiffness and cardiovascular risk.
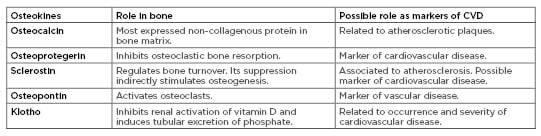
Table 1: Role of osteokines in bone metabolism and vascular disease.
CVD: cardiovascular disease.
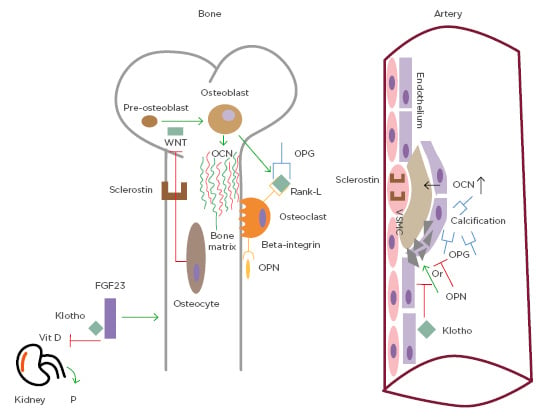
Figure 2. Common pathways of bone metabolism and vascular calcification.
Osteocalcin is the most important non-collagenous protein in the bone matrix. Elevated levels of osteocalcin have been associated with the presence of atherosclerosis. Osteoprotegerin prevents the interaction between receptor activator of nuclear factor Kb ligand and its receptor and its presence in the bloodstream is associated with vascular calcification. Sclerostin is a soluble factor released by osteocytes that inhibits the pathway of Wnt (which is important for osteoblast differentiation). It is correlated with atherosclerosis and present in vascular smooth muscle cells of the plaque. Osteopontin stimulates bone resorption, binding β-3 integrin and it is a marker of vascular calcification, while it is not clear if it prevents or stimulates it. Klotho is a co-receptor of FGF23, which inhibits renal activation of vitamin D and induces phosphate excretion. Decreased Klotho levels lead to increased calcification.
FGF23: fibroblast growth factor 23; OCN: osteocalcin; OPG: osteoprotegerin; OPN: osteopontin; P: phosphate; RANK: receptor activator of nuclear factor Kb; RANK-L: receptor activator of nuclear factor Kb ligand; VSMC: vascular smooth muscle cells.






