Abstract
Calcific aortic valve stenosis is the most common valve disease in the elderly population and is associated with significant morbidity and mortality. This condition is characterised by gradual fibrosis, thickening, and calcification of the affected leaflets, leading to decreased leaflet mobility and increased obstruction of the blood flow from the left ventricle. Lipoprotein(a) [Lp(a)] is a complex polymorphic lipoprotein with proatherogenic, proinflammatory, and prothrombotic properties. Several epidemiologic and clinical studies have described elevated Lp(a) levels as an independent causative risk factor for cardiovascular disease, including coronary artery disease, stroke, peripheral artery disease, heart failure, and venous thromboembolism. On the other hand, several studies have also described Lp(a) as a strong genetic causative risk factor for aortic valve calcification and aortic valve stenosis. In this review, the authors present and discuss the scientific and clinical evidence pertaining to the role of Lp(a) in calcific aortic valve stenosis.
INTRODUCTION
Lipoprotein(a) [Lp(a)] is a complex polymorphic lipoprotein synthesised by the liver. Lp(a) presents a similar structure with the low-density lipoprotein (LDL) molecule, only differing in the presence of the glycoprotein apolipoprotein(a) [(Apo(a)], which is covalently bound via a disulfide bond to the apolipoprotein B-100 (ApoB-100) of the LDL molecule.1 More specifically, Lp(a) comprises one molecule of an ApoB-100-containing LDL particle and one molecule of a large, highly polymorphic glycoprotein called Apo(a). A distinctive feature of Apo(a) is the presence of triple loop structures, which are called kringles. Kringle domains are stabilised by three internal disulfide bonds and are also found in certain coagulation factors, such as plasminogen, tissue plasminogen activators, prothrombin, and urokinase. However, in contrast to plasminogen, the linker sequences that join individual kringles are glycosylated in Apo(a). The two main components of Lp(a) are covalently linked together via a disulfide bond between the ApoB-100 of the LDL moiety and one of the kringle domains in Apo(a).2
Plasma levels of Lp(a) are under strict genetic control mainly by the LPA gene, largely unaffected by food intake, type of diet, presence of inflammation, or environmental factors.1,3 While the physiological role of the Lp(a) has not yet been well elucidated, this lipoprotein has been associated with several physiological processes, such as wound healing and tissue repair, as well as inhibition of cancer growth and spread.4 There is extensive evidence demonstrating that Lp(a) presents proatherogenic, proinflammatory, and prothrombotic properties, because it promotes the oxidation of LDL, enhances secretion and expression of proinflammatory cytokines, promotes platelet aggregation, and impairs plasminogen activation.5-8 Lp(a) is currently considered as an independent genetic, causative risk factor for cardiovascular disease (CVD), including coronary artery disease (CAD),9-11 stroke,10,11 peripheral artery disease,11 heart failure,12 and venous thromboembolism.13 In addition, several studies have also described Lp(a) as a strong causative risk factor for aortic valve calcification (AVC) and aortic valve stenosis (AVS).14-16
Calcific AVS (CAVS) is the most common valve disease in the elderly population, affecting >1 million patients in the USA, and is associated with significant morbidity and mortality.17 In Norway, the prevalence of AVS is also consistently increasing with age, average values being 0.2% in the 50–59 year old cohort, 1.3% in the 60–69 year old cohort, 3.9% in the 70–79 year old cohort, and 9.8% in the 80–89 year old cohort.18 In Sweden, the age-adjusted incidence of AVS declined from 15.0 to 11.4 in men and 9.8 to 7.1 in women per 100,000 from 1989–1991 and 2007–2009, and the median age at diagnosis increased by 4 years for both men and women.19 In a large systematic review and meta-analysis of population-based studies from 19 European countries and North America, the pooled prevalence of all AVS in the elderly (>75 years) was 12.4% and the prevalence of severe AVS was 3.4%.20 CAVS is characterised by gradual fibrosis, thickening, and calcification of the affected leaflets, thus leading to decreased leaflet mobility and increased obstruction of the blood flow from the left ventricle.17,21
The pathogenesis of AVS shares many similarities to that of atherosclerosis. Several longitudinal studies have demonstrated that hypercholesterolaemia has a significant impact on the development of degenerative AVS. Notwithstanding, in several clinical studies, intensive statin therapy has failed to halt the progression of CAVS or induce its regression.22-24 Here, it should be noted that statins have been shown to increase Lp(a) levels by roughly 10–20%,6,25,26 which could be one explanation as to why statins were proven ineffective in halting the progression of CAVS. In contrast, proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors have been shown to reduce Lp(a) levels by 24.5–29.5%.27 Related to this, it was recently shown that high levels of PCSK9 predicted development of AVS requiring surgery, although this association seemed to be driven by concurrent atherosclerotic disease.28
On the other hand, AVS hasalso been associated with several other atherogenic risk factors, such as diabetes, hypertension, smoking, male sex, as well as age, apparently due to progressive fibro-calcific remodeling.29 In addition, there are data indicating that chronic Chlamydia pneumoniae infection may act as a ‘trigger’ and aggravate AVS via the formation of circulating immune complexes. Importantly, a strong synergism was also observed between Lp(a) and C. pneumoniae IgG antibodies in circulating immune complexes.30 In this review, the authors aim to present and discuss the scientific and clinical evidence pertaining to the role of Lp(a) in CAVS.
STUDY EVIDENCE PERTAINING TO THE ROLE OF LIPOPROTEIN(a) IN AORTIC VALVE CALCIFICATION AND STENOSIS
There is extensive evidence from genetic and clinical studies demonstrating the causative association of elevated Lp(a) with CAVS.
Studies Establishing the Association of Aortic Valve Calcification and Stenosis with Increased Lipoprotein(a) Levels
The first report to demonstrate an association between Lp(a) and AVS was published in 1995. The study showed that AVS was present in 36.1% of subjects with Lp(a) levels ≥30.0 mg/dL, but only in 12.7% of subjects with Lp(a) levels <30.0 mg/dL (p<0.001).31
In another study, which evaluated the role of novel coronary risk factors in the development of AVC, it was clearly demonstrated that patients with AVC had significantly higher serum Lp(a) levels, as compared with subjects with essentially normal aortic valve morphology (27.4 mg/dL versus 19.9 mg/dL, respectively; p=0.033). Moreover, multivariate analysis identified Lp(a) as an independent predictor of AVC.32
Study Evaluating the Impact of Increased Lipoprotein(a) Levels on Aortic Valve Calcification in Patients with Familial Hypercholesterolaemia
Patients with familial hypercholesterolaemia (FH) have been shown to have a higher prevalence and extent of AVC than patients with non-familial hypercholesterolaemia.33 Exposure to classical risk factors alone cannot adequately explain the onset and progression of AVC in statin-treated FH patients.34 On the other hand, patients with FH have been shown to have 3-fold higher plasma Lp(a) levels than controls, apparently due to variation at the LDL receptor gene locus.35 Thus, in a study, which included 129 asymptomatic statin-treated patients with FH, and was designed to investigate whether Lp(a) concentration is associated with AVC in this cohort of patients, Vongpromek et al.34 showed that for a 10 mg/dL increase in Lp(a) concentration in those patients, there was an 11% associated increased risk of developing AVC. Multivariate analysis again identified Lp(a) as a significant independent predictor of AVC. Of note, however, Lp(a) levels were not associated with the presence or severity of coronary artery calcification.34
Genetic Studies
Genome-wide association studies (GWAS) are designed to test the association between markers, called single-nucleotide polymorphisms (SNP), across the genome and disease, usually involving ≥300,000 markers that are reasonably polymorphic and are spread across the genome fairly evenly.36 In other words, GWAS compare common genetic variants in large numbers of affected cases to those in unaffected controls to determine whether an association with disease exists.37 GWAS have been proven to be extremely useful in determining the association between elevated Lp(a) levels and CAVS.
In a GWAS, a SNP, rs10455872, in the LPA locus, encoding for Apo(a), was found to be significantly correlated with AVC (odds ratio [OR] per allele: 2.05). Importantly, the association between rs10455872 and Lp(a) levels was confirmed and it was clearly shown that Lp(a) levels mediate the effect of this SNP on AVC. Furthermore, in prospective analyses, LPA genotype was associated with incident AVS (hazard ratio [HR] per allele: 1.68) and aortic valve replacement (AVR) (HR: 1.54).14
In another study, which combined data from two prospective general population studies (the Copenhagen City Heart Study and the Copenhagen General Population Study), evaluating a total of 77,680 Danish individuals for as long as 20 years, elevated Lp(a) levels and corresponding LPA genotypes (rs10455872, rs3798220, kringle IV Type 2 [KIV-2] repeat polymorphism) were associated with an increased risk of AVS in the general population. More specifically, Lp(a) levels of 5–19 mg/dL, 20–64 mg/dL, 65–90 mg/dL, and >90 mg/dL were associated with multivariable adjusted HR for AVS of 1.2, 1.6, 2.0, and 2.9, respectively, as compared to levels of Lp(a) <5 mg/dL.15 Notably, the risk of AVS appears to increase continually as Lp(a) levels increase. In a large retrospective analysis, which assessed the correlation between Lp(a) levels and the incidence of AVS among patients with extremely high Lp(a) levels (>150 mg/dL), the prevalence of AVS was 3.74 times higher in the patients with Lp(a) levels >150.0 mg/dL, compared with those with Lp(a) levels <30.0 mg/dL.38
Furthermore, in a similar study, which used a multidirectional Mendelian randomisation approach and included 100,578 individuals from the general Danish population, elevated levels of Lp(a) were once again causally associated with increased risk of AVS. More specifically, a one-standard deviation (SD) increase in Lp(a) levels was associated observationally with a multifactorially adjusted HR of 1.23, whereas the corresponding causal risk ratios based on LPA SNP and on LPA KIV-2 genotype were 1.38 and 1.21, respectively. Of note, in this study, elevated Lp(a) levels were not causally associated with increased low-grade inflammation, as measured through levels of C-reactive protein (CRP).39
In another prospective Mendelian randomisation study, in which serum Lp(a) levels were measured in 17,553 participants of the European Prospective Investigation into Cancer (EPIC)-Norfolk study, elevated Lp(a) levels were associated with increased risk of AVS. More specifically, after adjusting for age, sex, and smoking history, participants in the top Lp(a) tertile had a 57% higher risk of AVS, as compared with those in the bottom Lp(a) tertile. Furthermore, heterozygotes and homozygotes for the rs10455872 genetic variant in the LPA locus were at increased risk for AVS with HR of 1.78 and 4.83, respectively, as compared to the individuals that did not carry the abnormal variant.40 These results were corroborated by a more recent, large case-control study, which replicated the association between LPA variants with AVS and showed a per risk allele OR of 1.34 for rs10455872 and 1.31 for rs3798220. Compared with individuals with no risk alleles, the homozygous OR for AVS was 2.05 for rs10455872 and 3.74 for rs3798220, while compound heterozygotes had a 2.00 OR for AVS.41
Studies Evaluating the Impact of Lipoprotein(a) and its Oxidised Phospholipid Content on Aortic Valve Calcification and Stenosis
In a study evaluating the impact of Lp(a) and oxidised phospholipids (OxPL) on ApoB-100 (OxPL-apoB), reflecting the biological activity of Lp(a), on AVS progression in 220 patients with mild-to-moderate AVS, elevated levels of Lp(a) and OxPL-apoB were associated with faster AVS progression and need for AVR. Based on the results of this study, the authors concluded that Lp(a) may mediate AVS progression through its associated OxPL.16 Of note, however, among patients with age ≤57 years, progression of AVS was 2-fold faster in those in the top Lp(a) tertile, as compared with those in the middle and bottom tertiles, whereas, in patients aged >57 years, the rate of AVS progression did not differ according to levels of Lp(a).16 This finding was corroborated in another more recent trial, in which for patients aged ≥70 years the development of AVS was not influenced by Lp(a) levels.42
In a secondary analysis of a randomised clinical trial, which included participants with mild-to-moderate CAVS with no indication for statin therapy from the ASTRONOMER (Effects of Rosuvastatin on Aortic Stenosis Progression) trial, the association of Lp(a) levels and its OxPL content with faster CAVS progression was actually linear. The results of this study reinforce the concept that measurement of Lp(a) levels should be performed in patients with mild-to-moderate CAVS to improve risk stratification and management.43
Studies Evaluating the Impact of Increased Lipoprotein(a) Levels on Aortic Valve Calcification and Stenosis in Patients with Bicuspid Aortic Valve
There is evidence suggesting that increased levels of Lp(a) are associated with the presence and severity of AVC in patients with bicuspid aortic valve (BAV). In an observational study, which looked at a small series of asymptomatic individuals with BAV, all the BAV subjects without AVC had normal Lp(a) levels, whereas 75% (3 out of 4) of cases with AVC had elevated Lp(a) levels. Although the author recognised that this was only a small series of BAV cases, notwithstanding, it was suggested that the concept that the plasma Lp(a) concentration may play a major role in the calcification process of the BAV would be worth exploring in larger samples.44 Later on, this concept was corroborated in another larger study, which investigated the association of Lp(a) and LPA KIV-2 repeat number with the presence of calcification and stenosis in patients with BAV. In that study, among BAV patients there was a clear positive association between Lp(a) levels and the degree of AVC. In contrast, lower LPA KIV-2 repeat numbers were observed in subjects with more severe AVC. Based on the results of this study, the authors suggested that Lp(a) may serve as a risk marker for the identification of BAV patients most likely to develop AVC and AVS.45 However, the potential effect of lowering Lp(a) on the development and progression of AVC in patients with BAV would need to be further investigated in larger randomised controlled trials.
Studies Addressing the Question Whether the Association of Elevated Lipoprotein(a) Levels with Aortic Valve Calcification and Stenosis is Dependent upon the Concomitant Presence of Coronary Artery Disease
As previously discussed, Lp(a) is considered an independent genetic, causative risk factor for CAD. Thus, the question may be raised whether the association of elevated Lp(a) with CAVS is dependent upon, or mediated by, the concomitant presence of CAD. In a nested, case-referent study, high Lp(a) levels and a high Apo B/A1 ratio were associated with surgery for AVS in patients with concomitant CAD, but not in those with isolated AVS (without concomitant CAD).46 However, in a more recent large genetic association study, genetically elevated Lp(a) levels were associated with CAVS independently of the presence of CAD, and individuals with high Lp(a) levels had a significantly increased risk for CAVS even in the absence of CAD. Based on the results of this study, the authors suggested that the measurement of Lp(a) levels in patients with CAVS might be proven clinically useful.47
POTENTIAL MECHANISMS THROUGH WHICH ELEVATED LIPOPROTEIN(a) MAY LEAD TO CALCIFIC AORTIC VALVE STENOSIS
As previously discussed, CAVS is the most common valve disease in the elderly population and its pathogenesis shares many similarities to that of atherosclerosis. Researchers have described an ‘early lesion’ that shared common histologic features with the early lesion of atherosclerotic plaques, suggesting that CAVS could be an atherosclerotic disease.21 Studies indicate that inflammation, lipid deposition, and fibrosis, which are all important contributors to atherogenesis, also play an important role in the pathogenesis and progression of CAVS.48,49 Lp(a) is a major carrier of proinflammatory OxPL.5 OxPL co-localise with Lp(a) in arterial and aortic valve lesions and thus may be directly involved in the pathogenesis of CVD and CAVS by promoting endothelial dysfunction, lipid deposition, inflammation, and osteogenic differentiation, leading to calcification. Thus, OxPL may potentially provide a mechanistic link between CVD and CAVS.50 In view of the similarities shared in the pathogenesis of atherosclerosis and CAVS, it can become easily understandable that molecules promoting inflammation and atherosclerosis, such as Lp(a), may also have a direct impact on CAVS.
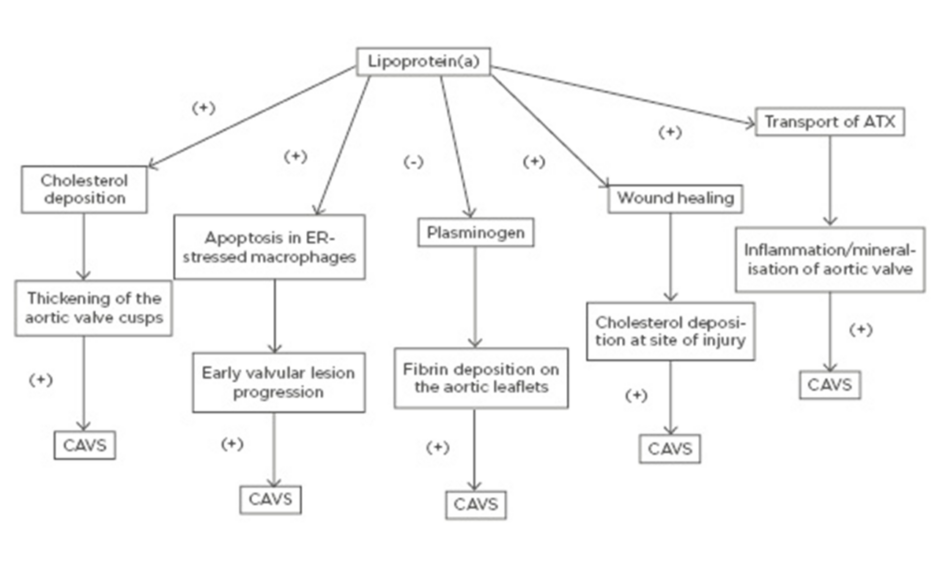
Even though the precise mechanism by which Lp(a) promotes the initiation and progression of CAVS has not been clearly elucidated, there are multiple proposed mechanisms through which elevated Lp(a) levels may lead to CAVS. Similar to LDL, transfer of Lp(a) from the circulation into the arterial intima and aortic valve cusps leads to cholesterol deposition with subsequent thickening of the aortic valve cusps.51
On the other hand, Lp(a) has been shown to trigger apoptosis in endoplasmic reticulum-stressed macrophages via a mechanism requiring both cluster of differentiation 36 (CD36) and toll-like receptor 2 (TLR2). This macrophage apoptosis may signify a key process, likely contributing to early valvular lesion progression.52,53
Furthermore, as it was mentioned earlier, Lp(a) exhibits prothrombotic properties because it competes with plasminogen and therefore prevents plasmin from dissolving fibrin clots,6-8 thus leading to fibrin deposition on the aortic leaflets and subsequent progression of AVS.51
Lp(a) is involved in the wound-healing process and may accumulate at sites of injury3 promoting cholesterol deposition. Thus, it may be well contemplated that accumulation of Lp(a) at sites of minor injury, such as the affected aortic leaflets at the very initial stages of AVS, may lead to increased cholesterol and thrombi deposition, thus promoting the progression of AVS.51
Finally, another molecule that has been implicated is autotaxin (ATX), a lysophospholipase D enzyme, which transforms lysophosphatidylcholine into lysophosphatidic acid (LysoPA). ATX is transported in the aortic valve via the bloodstream by Lp(a) and is also secreted by valve interstitial cells. ATX-LysoPA has been shown to promote inflammation and mineralisation of the aortic valve, thus promoting CAVS.54 More specifically, pericellular LysoPA may associate with lysophosphatidic acid receptor 1 (LPAR1), which promotes nuclear translocation of NF-κB. The activation of NF-κB leads to an increased expression of IL-6 and bone morphogenetic protein 2 (BMP-2), which are known pro-osteogenic factors.55
A schematic of the potential mechanisms through which elevated Lp(a) may lead to CAVS is shown in Figure 1.
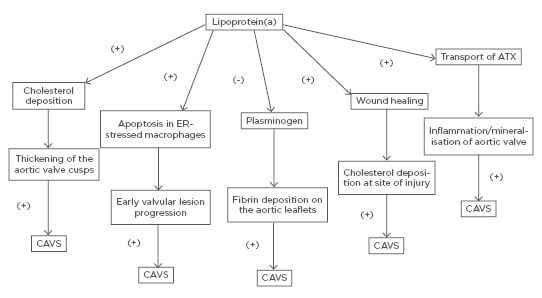
Figure 1: Potential mechanisms through which elevated lipoprotein(a) may lead to calcific aortic valve stenosis. ATX: autotaxin; CAVS: calcific aortic valve stenosis; ER: endoplasmic reticulum.
PRELIMINARY EVIDENCE PERTAINING TO THE EFFECT OF LIPOPROTEIN(a) LOWERING ON THE RISK OF AORTIC VALVE CALCIFICATION AND STENOSIS
In an elegant study, which used human aortic valve interstitial cells (HAVIC), it was clearly demonstrated, for the first time, that Lp(a) is causally involved in the induction of AVC. It was also shown that the LPA gene is locally expressed in the stenotic aortic valve. Lp(a) was found to induce osteogenic differentiation of HAVIC via induction of the gene encoding for the tissue-nonspecific alkaline phosphatase, as well as certain pro-osteogenic mediators. Moreover, it was shown that the Lp(a)-induced osteogenic differentiation of HAVIC was associated with an increase in the phosphorylation of several kinases implicated in cellular remodelling and apoptosis, such as mitogen-activated protein kinase-38 (MAPK38) and glycogen synthase kinase-3 beta (GSK3β). Inhibition of MAPK38 or GSK3β led to a significant reduction of Lp(a)-induced HAVIC calcification. Thus, interfering with the Lp(a) pathway could provide a novel therapeutic approach for the prevention or even reversal of CAVS.56
In another multimodality imaging study, Lp(a) and OxPL promoted valve calcification in patients with AVS. In this study, Lp(a) and OxPL-apoB levels were measured in 145 patients with AVS. Initially, on baseline 18F-sodium fluoride PET (18F-NaF PET), patients in the top Lp(a) tertile (>35.0 mg/dL) had increased valve calcification activity compared with those in the lower tertiles. Moreover, during follow-up, patients in the top Lp(a) tertile demonstrated increased progression of valvular CT-obtained calcium score, faster haemodynamic progression on echocardiography, and an increased incidence of AVR and death compared with those in the lower tertiles. Similar results were observed with OxPL-apoB. In vitro, Lp(a) induced osteogenic differentiation of HAVIC through its OxPL content. The Lp(a)-induced osteogenic differentiation of HAVIC was considerably attenuated with the E06 monoclonal antibody against OxPL. Again, these findings clearly demonstrate that elevated Lp(a) and OxPL-apoB levels promote AVC, and lowering Lp(a) or inactivating OxPL may potentially lead to slowing of AVC. Thus, these findings suggest that therapeutic approaches reducing elevated Lp(a) and OxPL levels in patients with AVS could slow disease progression and delay the need for AVR.57
Although to date no clinical trials have been conducted to assess the impact of Lp(a) lowering on the incidence of AVS, an analysis using data from a large prospective European cohort has shown that lowering Lp(a) to <50.0 mg/dL in the general population would be theoretically expected to reduce the overall incidence of AVS by 13.9%.58
CONCLUSION
The above review of the scientific, epidemiological and clinical data clearly demonstrates that Lp(a) plays an independent causative role in CAVS.However, additional evidence is needed to help us better understand the precise molecular mechanisms by which elevated Lp(a) causes CAVS and promotes its progression. Furthermore, it remains to be seen if pharmaceutical interventions that decrease Lp(a) levels would also be clinically effective in reducing the risk of CAVS and its associated morbidity and mortality.