Abstract
Lupus panniculitis occurs in 1–3% of the patients diagnosed with systemic lupus erythematosus (SLE) and 10% of the patients diagnosed with discoid lupus erythematosus (DLE). It is a disorder of autoimmune origin, manifesting as deep erythematous plaques and nodules involving the trunk, breasts, buttocks, face, and proximal extremities. It does not commonly ulcerate. This report highlights the case of a 22-year-old Asian female with a history of coeliac disease and significant family history of antiphospholipid antibody syndrome (APS) who presented with fever, malaise, weight loss, and subcutaneous non-tender nodules over the forearm, back, bilateral thighs, and feet. Laboratory investigations revealed positive antinuclear antibodies, anti-Ro/SSA antibody, and lupus anticoagulant, resulting in a diagnosis of APS. Biopsies of lesions were consistent with findings of lupus panniculitis. Every case of SLE and DLE with discrete skin lesions should be reviewed for any distinct entity such as lupus panniculitis, as it may be associated with greater risk of flares and systemic involvement. The purpose of this case report is to emphasise that early diagnosis and prompt treatment is crucial to improving the prognosis of such patients.
INTRODUCTION
Lupus panniculitis, also known as lupus profundus,1 is characterised by chronic and recurrent inflammatory lesions of subcutaneous tissue.2 It mostly occurs as a primary disorder but could also be associated with underlying systemic involvement in discoid lupus erythematosus (DLE).3 The terminology lupus panniculitis was first coined by Kaposi4 in 1883. It usually occurs in the age range of 20–60 years old with a female predominance of 2:1.2,5 It presents as a firm, fixed, hard skin lesion that may or may not be tender.6 The diagnosis is usually performed by clinical correlation, histological evidence, immunofluorescence, and autoimmune profile.6 Systemic lupus erythematosus (SLE) is an autoimmune disorder that could potentially involve any organ or system.7 Cutaneous lupus erythematosus, usually a benign disease, can become complicated in 1–3% of patients and progress into lupus profundus.8
CASE PRESENTATION
A 22-year-old female of Asian descent with a past medical history of coeliac disease (CD) and positive family history of anti-phospholipid antibody syndrome (APS) in a second-degree relative presented with fever, generalised weakness, miscarriages in two previous pregnancies, and unintentional weight loss. She also presented with arthralgia, alopecia, photosensitivity, heavy menstrual blood flow, bluish discoloration of the fingers on exposure to cold, and recurrent oral ulcers. She developed non-tender subcutaneous nodules that formerly involved the forearm and later involved the back and bilateral thighs and feet. She denied facial rash, easy bruising, seizures, psychosis, proximal muscle weakness, cough, chest pain, diarrhoea, and blood in sputum. She was treated empirically with intravenous paracetamol (1 g every 8 hours) and corticosteroids (hydrocortisone 100 mg, every 8 hours).
The physical examination was unremarkable except for mild pallor; swelling in wrists, ankles, and small joints of the hands; periorbital puffiness; oral ulcers; non-scarring alopecia; and non-tender subcutaneous nodules over the forearm, back, and bilateral thighs (Figure 1). Laboratory workup showed haemoglobin: 10.2 g/dL with mean corpuscular volume: 70 fL; total leucocyte count: 9×103 u/L; platelets: 30×103 U/L; total bilirubin: 0.86 µmol/L with direct component of 0.23 µmol/L; alanine transaminase: 84 U/L, aspartate transaminase: 65 U/L, alkaline phosphatase: 140 IU/L; and gamma-glutamyl-transferase: 113 U/L. Other baseline investigations such as erythrocyte sedimentation rate and C-reactive protein were within normal limits. Based on suspected history, an autoimmune profile was sent which showed positivity for antinuclear antibodies (ANA) (2+, granular pattern), anti-dsDNA (40 U/mL), anti-Ro/SSA antibody (14 U/mL), and lupus anticoagulant antibody, whilst anti-La and other antibodies panel were negative, along with normal complement levels. The differential consideration at this point included SLE, mixed connective tissue disease, APS, Sjogren syndrome, rheumatic fever, and hypothyroidism. Based on blood workup, the initial diagnosis of SLE with APS was made and the patient was scheduled for skin-biopsy for the subcutaneous nodules.
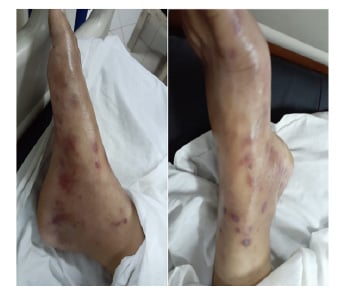
Figure 1: A patient presenting with non-tender subcutaneous nodules over the feet.
Biopsy of the skin nodule showed lobular panniculitis, hyaline degeneration of fat, and lymphocytic infiltrate with scattered plasma cells, with no epidermal atrophy and no evidence of vasculitis (Figure 2). Immunofluorescence studies demonstrate deposits of IgM and complement 3 (C3) along the dermoepidermal junction, confirming the diagnosis of panniculitis in association with APS on a background of SLE. However, no specific feature suggestive of APS was present in the skin biopsy. The patient was managed initially with steroids (methylprednisolone 20 mg, twice daily), and hydroxychloroquine (HCQ) in a dose of 200 mg, every 12 hours. The general condition improved within a week; the patient was discharged on oral corticosteroids (1 mg/kg) and mycophenolate mofetil (500 mg, twice daily), and they were followed-up in the ambulatory setting within 2 weeks with a much-improved condition. Anticoagulant (warfarin 5 mg every 24 hours) was then given at the outpatient follow-up after 2 weeks. The patient was continued on the medications and was scheduled for monthly out-patient follow-up.
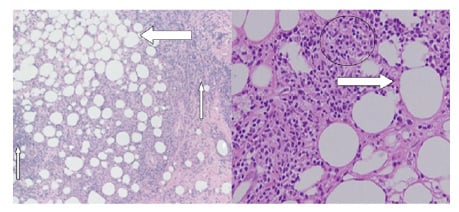
Figure 2: A) The hyaline degeneration of fat (thick arrow) with lymphocyte infiltration and plasma cells (thin arrows). B) The lobular panniculitis, fat degeneration (marked with an arrow), and inflammatory cells (marked with a circle).
DISCUSSION
Lupus panniculitis is characterised by the development of painful indurated dermohypodermal nodules or plaques, typically affecting the thighs, the upper arms, or the cheek area of the face.9 Bednarek et al.10 reported in their study that in patients diagnosed with SLE, only 1–3% develop lupus panniculitis, while in patients diagnosed with DLE, this number can increase up to 10%.2
The terminology ‘lupus panniculitis’ was first introduced by Kaposi4 in 1883. The disease has a female to male ratio of 2:1, occurring predominantly in the age group of 20–60 years old.5 The illness manifests itself as deep erythematous plaques and nodules, involving the trunk, breasts, buttocks, face, and proximal extremities. It does not commonly ulcerate. The lesions may or may not be tender, depending upon the severity, and often heals with scarring and atrophy which can lead to significant cosmetic disfigurement.11 The presentation is mostly of firm, fixed, hard lesions, which have the propensity to involve the fatty tissue, ultimately leading to calcification or atrophy of the lesion.12
In patients with such a presentation, other inflammatory disorders such as factitial panniculitis, traumatic panniculitis, morphea profundus, dermatomyositis, and subcutaneous panniculitis-like T cell lymphoma should be considered and ruled out. Amongst these, the greatest mimic is cutaneous T cell lymphoma, though it does need to be kept in mind as it has an aggressive course and involves poor prognosis.13 The diagnosis of lupus panniculitis involves clinical correlation, histological evidence, immunofluorescence, and autoimmune profile.14 In approximately 50% of cases a biopsy of the specimen leads to the diagnosis, revealing the typical changes such as perivascular and peri-appendageal lymphocytic inflammation, hydropic degeneration of basal layer, hyperkeratosis, thickened basement membrane, interface changes, epidermal atrophy, and hyaline sclerosis of lobules.15 Immunofluorescence studies are also an important subset helping in the diagnosis; they display the linear deposits of IgM and C3 along the dermoepidermal junction. If the biopsy gives equivocal results, the lupus band test is often performed. However, the ultimate diagnosis requires the clinical background with these pathological features and positive ANA occurs in about 50–85% of patients.14 In patients who test ANA negative, the diagnosis should solely rely on histopathological evidence.15 Lupus pannulitis has been rarely reported with APS, and usually without any systemic involvements.16 Also of note, eyelid oedema is the most common ocular involvement reported with lupus panniculitis.17
The 22-year-old Asian female, a known case of CD with a significant family history of APS, presented with fever, malaise, and weight loss. Her medical history was significant for oral ulcers, alopecia, Raynaud’s phenomenon, and recurrent miscarriages. She had subcutaneous non-tender nodules located on the arms, thigh, and buttock region. Laboratory investigations revealed positive ANA, anti-Ro/SSA antibody, and lupus anticoagulant, resulting in a diagnosis of APS on a background of SLE. Biopsies of lesions were consistent with findings of lupus panniculitis, ultimately leading to a diagnosis of lupus panniculitis in association with APS, on the background of SLE. The patient was given steroids (methylprednisolone, 1 mg/kg) along with mycophenolate mofetil (500 mg twice daily) and was followed-up in the ambulatory setting within 2 weeks. Anticoagulant (warfarin, 5 mg daily) was added to the patient prescription at the outpatient follow-up after 2 weeks.
The treatment of lupus panniculitis is challenging because there are no specific indications or medication recommendations in the guidelines.18 Antimalarial drugs are believed to bring improvement in mild cases of isolated lupus panniculitis.19 Chloroquine in a dose of 200–250 mg a day and HCQ in a dose of 200–400 mg a day is typically recommended. Usually, an eye examination is carried out during HCQ therapy in the first year if forced by a higher ophthalmolotoxic profile, but the risk remains small in therapeutic doses. HCQ is being used in SLE patients with mild disease and is known to decrease the need for steroids.18
When lupus panniculitis co-occurs with SLE, a combination of antimalarial and steroids is prescribed. Systemic steroids are reserved for aggressive and resistant cases.11 In association with APS, lifelong anticoagulant therapy is usually added to the empiric treatment to prevent the recurrence of thromboembolism.20 Surgical intervention is indicated when all other possible approaches have failed to treat lupus panniculitis.20
CONCLUSION
Every case of SLE and discoid lupus with discrete skin lesions should be reviewed for any distinct entity such as lupus panniculitis, as it may be associated with greater risk of flares and systemic involvement. The purpose of this case report was to emphasise that early diagnosis and prompt treatment is crucial to improving the prognosis of patients with lupus panniculitis. It is considered the least common variety of cutaneous manifestations of lupus erythematosus, but it can be considered an initial presenting manifestation of SLE.