BACKGROUND AND AIMS
This abstract outlines the case of a 57-year-old Georgian woman referred to an allergist by a gastroenterologist due to a 10-day history of sudden-onset cramping, left lower quadrant abdominal pain, diarrhoea, and weight loss, accompanied by generalised oedema. The patient experienced an increased attack frequency of 20–25 episodes per year, occurring randomly without discernible triggers and typically resolving within 3–5 days. Symptomatic treatment with H1-antihistamines proved ineffective. The patient’s medical history revealed symptom onset at 2 years old, with an unspecified first episode, and at 8 years old she underwent bowel resection for diagnosed bowel obstruction. Significantly, hereditary angioedema was prevalent in the family, affecting her brother (fatal laryngospasm at 25 years old), grandfather, two uncles, and a cousin.
MATERIALS AND METHODS
For this study, the authors conducted complement studies of C4 protein levels, C1 esterase inhibitor concentration and function, and total IgE level (Table 1).
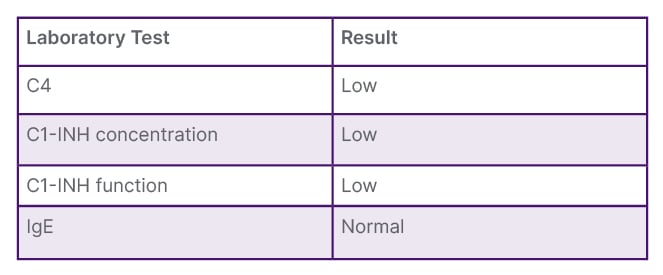
Table 1: Complement studies: C1-INH concentration, C1-INH function, C4, and total IgE.
C1-INH: C1 esterase inhibitor.
RESULTS
Consequently, the patient was diagnosed with hereditary angioedema (HAE) Type I (Table 2). Management involved a routine medication regimen of danazol at 200 mg three times a day. The dosage was titrated to the usual maintenance dosage, supplemented by emergency guidelines.
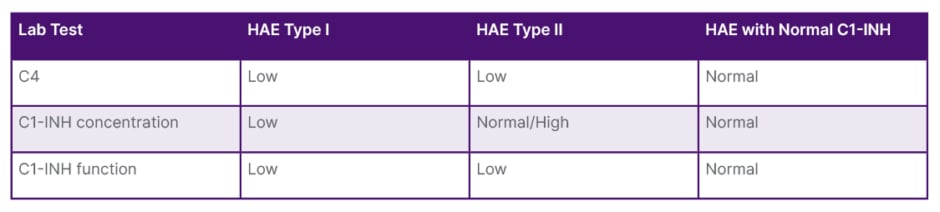
Table 2: Classification of the different forms of hereditary angioedema.
C1-INH: C1 esterase inhibitor; HAE: hereditary angioedema.
CONCLUSION
This abstract highlights the diagnostic complexities associated with the often non-specific symptoms of hereditary angioedema, underscoring the need for a thorough medical history and meticulous laboratory assessments for precise identification and effective management of this challenging condition.
Key takeaways include that HAE is characterised by recurrent, genetically mediated angioedema associated with increased vascular permeability primarily due to bradykinin; common HAE attack locations are the face, throat, abdomen, hands, feet, and genitals; and that attacks may be triggered by injury, stress, or even excitement, but may also appear for unknown reasons.